Tacrolimus, astemizole and a new screening approach for drugs that reduce PrP
 Read with caution! This post was written during early stages of trying to understand a complex scientific problem, and we didn't get everything right. The original author no longer endorses the content of this post. It is being left online for historical reasons, but read at your own risk. |
| Tacrolimus | Astemizole |
 |
 |
A few hours ago someone left me a comment (thanks!) pointing me to a new prion therapeutic study hot off the press in PNAS from the folks at Scripps Florida: Karapetyan & Sferrazza 2013, not even indexed by PubMed yet.
I’m fairly excited about this study, mostly because it represents a new screening approach that looks promising. The authors explain the rationale for their approach in a lengthy intro. Observing that knockout mice are resistant to prion disease [Bueler 1993 and others] and that abolishing PrP altogether can reverse symptoms in prion-infected mice [Mallucci 2003], the authors were interested in developing an assay to discover compounds that simply reduce PrP levels, in contrast to other investigators’ approaches (for instance the Ghaemmagami and Groschup labs both screen for compounds that reduce PrP-res formation in infected cells). The authors further note that cleaving PrP’s GPI anchor to release the protein from the cell surface can completely abolish prion infection in cell culture [Enari 2001], so they reasoned that what’s important is not total PrP levels across the cell, but rather PrP levels at the cell membrane. Their vision, then: an assay that detects levels of PrP on the cell surface.
One advantage of this approach is that it doesn’t involve PrPSc. You can just screen in ordinary, uninfected, wild-type cells, which makes it easier to adapt to high-throughput techniques – presumably, a lot less hassle with biosafety and so forth, though the authors don’t go into detail. The authors wanted an assay that creates an optical readout of PrP levels – which usually means antibodies – but wanted to avoid the extra step of washing off unbound antibody (which is a step in ELISA assays, such as those used in the Ghaemmagami lab). Therefore they adapted an existing technique called Fluorescence Resonance Energy Transfer (FRET).
FRET is fascinating, but tl;dr: attach two different antibodies to PrP at different epitopes (locations on the protein), each conjugated to a different fluorescent element with a different wavelength (say, blue fluorescent protein and green fluorescent protein). Excite one of them, and if-and-only-if it is connected to the other, the other one will emit its wavelength of light back at you. Thus you only get a signal if both antibodies are bound to the same molecule. That means you don’t need to wash off unbound antibody – any antibody that is unbound it just won’t emit any light at you.
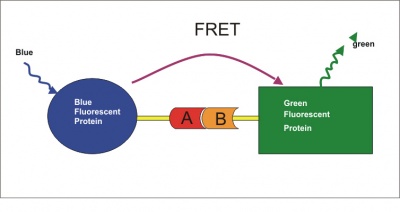
(Diagram by Paul Held)
Following the initial FRET screen, the authors also did a number of other experiments to clarify the mechanism of action of their hits – details in supplement.
One thing I find a bit confusing. The authors note that their assay would detect PrP that had been shed from the cell surface into the culture medium, and that to get around this problem they used LD9 cells which exhibit minimal shedding of PrP. It seems to me that this would prevent you from picking up hits from any compounds whose mechanism of action is to cleave the GPI anchor, such as glimepiride [Bate 2009]. Am I wrong?
Karapetyan & Sferrazza applied their new assay to mouse neuroblastoma (N2a) cells with a library of already-FDA-approved drugs: MSDI’s 1,280-compound US Drug Collection. There were nine hits, including tannic acid, tacrolimus and astemizole, the latter two of which they flagged for followup. A counterscreen showed that both of them reduced PrP-res formation in prion-infected neuroblastoma cells. Tacrolimus reduced PrP both at the cell surface and intracellularly, but didn’t affect PrP mRNA, thus they reasoned it must either inhibit PrP translation or promote PrP degradation. Curiously, astemizole was found not to have significantly reduced PrP mRNA nor protein levels, whether at the cell surface or inside the cell. That’s odd because the whole point of their FRET assay was to detect compounds that reduce PrP levels at the cell surface. The key here is that they did the protein and mRNA quantification at a lower concentration of astemizole (2μM) which had proven effective in the PrP-res counterscreen but was much lower than the screening concentration. So presumably astemizole does reduce PrP protein levels at the surface at a higher concentration, but not detectably so (yet for some reason it still inhibits PrP-res formation) at a lower concentration. Instead, astemizole was found to increase autophagy, and that is the presumed mechanism of action for its effect at 2μM.
The authors then sought to validate these two compounds in mice. I find their mouse study a bit surprising: mice were intracerebrally infected with RML prions, then treated with the drugs from 20 to 50 days post infection (dpi). The authors note that “Intracerebral prion infection combined with a short i.p. drug application represents a stringent model allowing detection of only the most effective antiprion drugs.” No doubt that i.c. infection and i.p. treatment are the way to go, yet I find the treatment specifically from 20 to 50 dpi to be both too stringent (you’ll miss compounds that might have worked if administered continuously straight through the disease course) yet also not stringent enough (20 dpi is very early, corresponding to a point in disease course in humans when no sporadic CJD patient will yet have a diagnosis or be enrolling in a clinical trial). In any event, control mice died after 149 days, and the astemizole-treated mice lived an average of 155 days. That may represent a 4% increase in survival time – but the statistical significance is marginal (p = .06). Further work will be needed to confirm that the effect is real. The tacrolimus mice were no different from controls.
Let’s get to know these drugs.
Tacrolimus (aka FK-506) was discovered in 1984, FDA-approved in 1994, and is now available as a generic. It’s an immunosuppressant used in organ transplants and for a few other things. It acts on the mTOR pathway like rapamycin but its exact mechanism is different – it binds FKBP12 and whereas rapamycin inhibits B and T cells’ response to IL-2, tacrolimus inhibits T cells’ production of IL-2. Its pharmacokinetics [reviewed in Venkataramanan 1995] combined with the high dose required for prion inhibition according to Karapetyan & Sferrazza’s results apparently mean it will be hard to achieve therapeutic levels in the brain. Karapetyan & Sferrazza used 1.5 mg/kg/day in mice, which corresponds to about 0.12 mg/kd/day in humans, which is within the range used clinicall: drugs.com gives (oral) doses no more than 0.2 mg/kg/day in humans. But as you can imagine, since it’s an immunosuppressant the side effects are not exactly things you want.
Astemizole has a colorful history. It was discovered in 1977, used as an antihistamine for years (it’s an H1 antagonist) and then withdrawn from the market in 1999 because it very rarely had a side effect of cardiac arrythmia and, after all, by then a new generation of antihistamines had come out. Despite being with drawn, astemizole is in the scheme of things a very well understood drug that is quite safe and (according to Karapetyan & Sferrazza) still sold in many countries. Also it actually delayed disease in the mice. The observed delay in disease for the mice was tiny (155/149 dpi = 4% delay at p = .06), but since the treatment duration was so much briefer than many other studies have used, I feel that I still don’t have any good basis to compare its efficacy to that of the many other drugs I’ve reviewed on this blog. It’s hard to find dose information on astemizole since it’s not on the market in the U.S. anymore, but rxlist.com seems to suggest that 10mg/day was a typical dose; the mice received 3 mg/kg/day (*(3/37)*70) = ~17mg/day for a 70 kg human.
At this point, I’m not sure how much stock to put in these two drugs. They could yet prove to be fairly effective, particularly if used in a cocktail with other drugs that reduce PrP levels.
But without putting too much faith in these specific two drugs, I was heartened by Karapetyan & Sferrazza’s paper. I find the evidence pretty convincing that reducing or eliminating PrP is the best strategy for fighting prion diseases (discussed a bit in this gene therapy post, and more investigation of this topic in a forthcoming post), so it is exciting news that these folks have designed an assay specifically aimed at this outcome.
update 2013-04-04: I just spoke with Dr. Corinne Lasmézas, the last author on the study, who generously took some time to chat with me about this study and her lab’s future plans (thanks!). She answered a couple of my questions about the paper and gave me permission to write about her answers here:
- As discussed earlier in this blog post, it seems to me that this assay would have little or no power to detect drugs whose mechanism of action is to promote cleavage of the GPI anchor. Dr. Lasmézas agreed that is probably true. The assay is, however, powered to detect drugs that act at several different stages: transcription (see #2 below), translation, passage through the secretory pathway, or degradation.
- In the present study [Karapetyan & Sferrazza 2013], the cells were incubated with the drugs for 24h (stated in supplement) before antibodies were applied. Dr. Lasmézas noted that the half-life of PrP protein is 3-6h, so a 24h incubation is plenty of time for a complete turnover. I noted that in Tremblay 1998, the first study to place PrP under a Tet-off switch, it was observed that PrP levels did not reach their minimum until the mice had been treated with doxycycline for one week. I had taken this to mean that PrP mRNA has a fairly long half-life and therefore that this 24h incubation time may not have been adequately powered to detect drugs that inhibit PrP transcription. Dr. Lasmézas pointed out that this finding in the Tet-off mice may have been due to pharmacokinetics in vivo and that mRNA suppression may well be much faster in cell culture. Neither of us were aware of any studies that have specifically examined the half-life of PrP mRNA**. She also pointed out that the screen would either way detect drugs that act to destabilize PrP mRNA. **update 2013-05-07: turns out [Pfeifer 1993] quantified PrP mRNA half life at 7h in both N2a and ScN2a cells; see half-life post
In sum: the assay is probably not ideal for detecting drugs that cleave the GPI anchor, might or might not be optimal for transcription inhibitors, and is definitely well powered to detect a variety of other mechanisms of action that reduce PrP levels.
update 2013-05-13: It has been pointed out to me that both of these drugs had gained attention previously as potential therapeutics. Astemizole turned up in a screen a decade ago [Kocisko 2003], though its mechanism was not investigated at the time, and so it was not reported that it reduced total PrP. It was later used as a (weak) positive control in another screen [Bertsch 2005]. Tacrolimus has been tested successfully in mice – it increased survival even when administered at onset of clinical signs, apparently due to calcineurin inhibition rather than by reducing PrP levels [Mukherjee 2010].