How do prions travel from cell to cell?
In his talk at Prion2013, Andy Hill mentioned that there were three known mechanisms by which prions can travel from cell to cell: direct cell-cell contact, tunneling nanotubes, and exosomes. The aim of this post is to get familiar with each of these mechanisms.
tunneling nanotubes
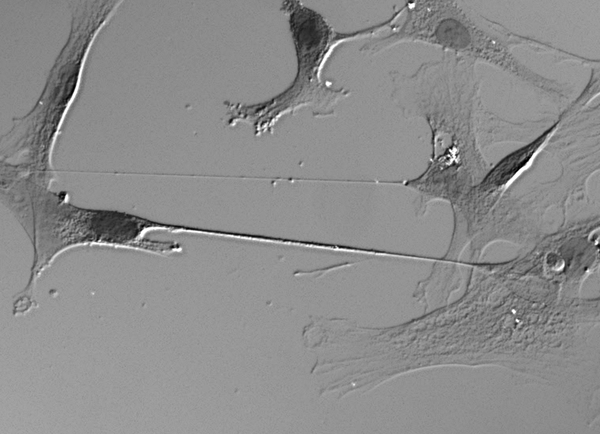
Above: tunneling nanotubes (two lines across the center) between human ells in culture, from Lou 2012, Fig 1e.
Tunneling nanotubes, or TNTs, are a relatively new thing in our understanding of biology, having been discovered just a decade ago [Rustom 2004]. They’re membrane-bound tubular passageways that form between adjacent cells. It starts with a filopodia-like finger stretching out from one cell until it touches the other. The tubes are 50 to 200nm in diameter [Rustom 2004] and can be 10 to 80 μm long [Gousset & Zurzolo 2009], and contain an actin filament used to transport membrane-bound vesicles and organelles while not allowing many small molecules through. TNTs appear to represent a new form of intercellular communication.
And, it appears that TNTs are also a route for prions to move from cell to cell [Gousset 2009]. The authors’ review of their own work in Prion is a good read, especially if you don’t have a route past Nature’s paywall [Gousset & Zurzolo 2009]. In general, PrP moves around intracellularly on membrane-bound vesicles [Encalada 2011] and after prion infection, PrPSc gets taken up into these vesicles [Magalhaes 2005], so it seemed natural to ask whether PrPSc in vesicles might be able to traverse TNTs. Gousett found that PrPSc could indeed travel intercellularly via TNTs, and this was confirmed in multiple different neuronal and non-neuronal cell lines. Not only could TNTs be one way that PrPSc moves between neurons, they also appear to be a potential way for PrPSc in peripheral prion infection to move from dendritic cells to peripheral neurons on their way to neuroinvasion.
exosomes
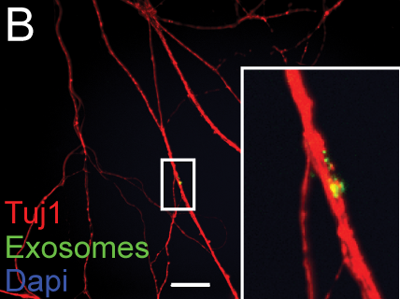
Above: exosomes (green) being taken up by the axon (red) of a neuron. From Fruhbeis 2013, Fig 7b.
Exosomes are tiny membrane-bound vesicles released from cells. Scientists have known about them for much longer than TNTs – according to Wikipedia, the earliest report of them is [Johnstone 1987]. They are round, with a typical diameter of ~100 nm, and contain proteins and RNA, particularly miRNA. Their roles in intercellular signaling are still being studied.
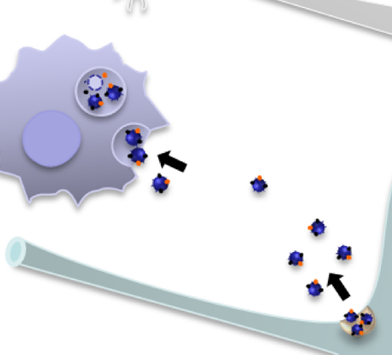
Above: exosomes (dark blue) traveling from cell to cell. From Fruhbeis 2013, Fig 9.
The way that exosomes are released and uptaken is, to me at least, a bit non-intuitive. Consider that in endocytosis, a bit of the cell’s membrane buds in, forming a membrane-bound vesicle of extracellular material floating in the cytosol. Therefore by analogy you might assume that exosomes involve a bit of the cell’s membrane budding off the other direction, leaving a little membrane-bound bubble of cytosoling material floating extracellularly, and that exosomes would be taken up by fusing with the membrane of the destination cell.
That’s not quite how it works. Instead, as shown in the above image, exosomes bud off from the inside of endosomal vesicles so that now you have little bubbles of cytosol floating inside a bubble of extracellular material floating inside the cytosol. These structures are called multivesicular bodies (MVBs). When these fuse to the cell surface, the exosomes are freed into the extracellular space. Symmetrically, when exosomes reach their destination they’re taken up into endocytic vesicles, not directly into the cytosol from the cell surface.
PrP is abundant on exosomes – so much so, said Andy Hill, that you could almost use PrP as a marker of exosomes. Regardless of whether they’re prion-infected or not, cells release loads of PrP on exosomes [Fevrier 2004]. When you maintain cells in culture, the culture medium ends up full of exosomes. In fact, many (most?) biological fluids are loaded with exosomes – that includes everything from urine [Gonzales 2010] to the fetal calf serum used in research labs (that’s why they specially sell exosome-depleted FCS). So Fevrier used a clever trick to see if exosomes could transmit prion infection: the culture medium from infected cells was pelleted and then resuspended and dumped onto uninfected cells, which subsequently became infected. The fact that the two populations of cells never touched each other rules out a mechanism like cell-to-cell contact or TNTs.
But that alone doesn’t absolutely indict exosomes – it still leaves the possibility of shed PrP or extracellular aggregates of PrP as a source of infectivity in the culture medium. Fevrier was able to link the infectivity to the floating fraction from the culture medium, which might have been a form of evidence but I don’t fully understand it (do exosomes float but pure PrP doesn’t??). A later study performed fractioning of the medium and demonstrated that infectivity was highest in the fractions containing exosomes, which might be the most solid piece of evidence for PrP infectivity traveling via exosomes [Coleman 2012 (ft)]. As an aside, it’s been suggested that the PrP in exosomes is a particular sub-population of PrP molecules – it’s depleted for unglycosylated PrP and, strangely, it doesn’t react with N-terminal antibodies yet appears to be full-length [Vella 2007].
cell-to-cell contact
Direct contact between cells was actually the first reported mechanism for intercellular prion transmission [Kanu 2002]. Kanu mixed prion-infected SMB cells with uninfected HMH1 cells and grew them together for a week, during which time the two cell types formed “a closely packed monolayer”. Then the SMB cells were killed off with an antibiotic that only the HMH1 cells had resistance to, and the HMH1 cells were cultured further and then assayed for PK-resistant PrPSc. Sure enough, they had become infected. When Kanu repeated the experiment with the two cell types separated by a porous surface, no PrPSc was transferred, even though the pores were large enough (400 nm) for exosomes to fit through. The paper doesn’t discuss whether the cell populations were too far apart for TNTs, because TNTs wouldn’t even be discovered until 2 years later. Thus it’s not clear whether this experiment really proves direct cell-cell contact or if prions could have been spreading via TNTs when the cells were co-cultured.
In a second experiment, Kanu grew live uninfected cells in the presence of aldehyde-fixed, dead, prion-infected cells, and the live cells became infected. That experiment is a bit more convincing that active transport along TNTs was not involved, though it’s still possible that the wandering filopodia of the live cells picked up a prion infection from their dead neighbors. Moreover, another study on different cell lines [Paquet 2007 (ft)] found that very little prion infectivity was transferred from dead cells to live cells.
other mechanisms?
Like Kanu, Paquet found that cells could be infected by co-culturing with live infected cells. Unsurprisingly, the cells adjacent to the infected cells were the most likely to become infected. TNTs were not discussed as a mechanism, but Paquet did propose a few possible mechanisms, including “cell painting” in which GPI-anchored proteins can leave one cell’s surface and become embedded in another cell’s surface. Paquet noted that one sketchy thing about proposing that PrPSc floats directly from cell to cell is that PrPSc isn’t released from the cell surface by PIPLC, an enzyme which cleaves the GPI anchor [Caughey 1990, Stahl 1990]. To me, it’s not clear whether proteolytic shedding (which is distinct from GPI anchor cleavage) could still play a role in releasing PrPSc from the surface. Anchorless PrP has a way of forming extracellular plaques, and co-expression of anchored PrP makes the phenotype even worse [Stohr 2011], which makes me wonder whether anchorless (or shed) PrP can help spread prion infection from neuron to neuron.
Another possibility is microvesicles [Mattei 2009]. The Wikipedia entry on microvesicles was unhelpful to me because it considers the term “microvesicles” to refer to all membrane-bound vesicles released from a cell, including exosomes. From looking at Mattei’s paper it appears that they are referring specifically to microvesicles formed by direct budding off of the plasma membrane into the cytosol. (This, unlike the process by which exosomes are formed, is a mirror image the process of endocytosis).
discussion
There are a number of therapeutic strategies to slow the spread of prions from the periphery to the brain [Mabbott & MacPherson 2006] but from looking at these mechanisms, there doesn’t appear to be an obvious therapeutic route to slowing prion spread within the brain. However, the subject of cell to cell spread of prions is still quite interesting from the standpoint of prion kinetics.
How quickly do prions spread from cell to cell? For cell culture models, I see conflicting data from co-culture experiments. Kanu 2002 reported that a high percentage (10-30%) of cells could be infected by 7 days of co-culture, but another experiment reported a negligible fraction of cells becoming infected even after 35 days of co-culture [Ghaemmaghami 2007], concluding that almost all prion transmission in cell culture is vertical, i.e. from mother to daughter cells. The difference between Kanu’s and Ghaemmaghami’s results could result a difference in cell lines or in experimental techniques. Overall, though, the fact that cultured cells are generally so darn hard to infect and that even huge titers of prions only infect ~10% of cells [Klohn 2003] suggests that in most cell lines people have studied, the horizontal spread of prions is relatively slow.
But all of the studies reviewed here are from cell culture experiments – it’s harder to imagine how you’d watch the spread of prions in vivo. But for all we know, the kinetics could be quite different in the live brain.
By the end stages of prion disease, PrPSc is found in all parts of the brain [Safar 2005a], but how quickly does it get there? Prion strains have particular patterns of neurotropism and (as far as I’m aware) this doesn’t depend on where the initial prion inoculation occurs. Yet we know that PrPSc accumulation and prion neurotoxicity are time-dependent phenomena, so why wouldn’t the neurons exposed first to PrPSc, right at the inoculation site, be the first to go?
One possibility is that cell-to-cell spread of prions in vivo is quite rapid, so that infectivity spreads across the brain within, say, the first several days of prion infection and that from then on, all neurons have “equal opportunity” to accumulate PrPSc according to their availability of strain-specific co-factors or compatible PrPC glycoforms, etc.
Another possibility is that there is some systemic protective mechanism at work (something that all those reactive glial cells are doing? PrPC downregulation?) that limits PrPSc accumulation and/or neurotoxicity in the early-infected neurons until it becomes overwhelmed by a brain-wide prion infection. Yet it is hard to reconcile this view with the cell-autonomous nature of prion neurotoxicity, specifically with the fact that an infected graft can degenerate even in an otherwise healthy brain [Brandner 1996].
Any other ideas? Leave me a comment.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.