Proteolytic shedding of prion protein
shedding
“Shedding” of PrP refers to release of PrP from the cell surface. This can be achieved experimentally with PIPLC, an enzyme that cleaves GPI anchors, but I haven’t read any reports that cleavage of PrP’s GPI anchor is a mechanism for shedding in vivo. Instead, as we’ll see, shedding appears to result from cleavage of PrP itself, at a peptide bond near the C terminus. Here’s a diagram of these two phenomena. As I’ll explain later, the exact cleavage site in human PrP is not known.
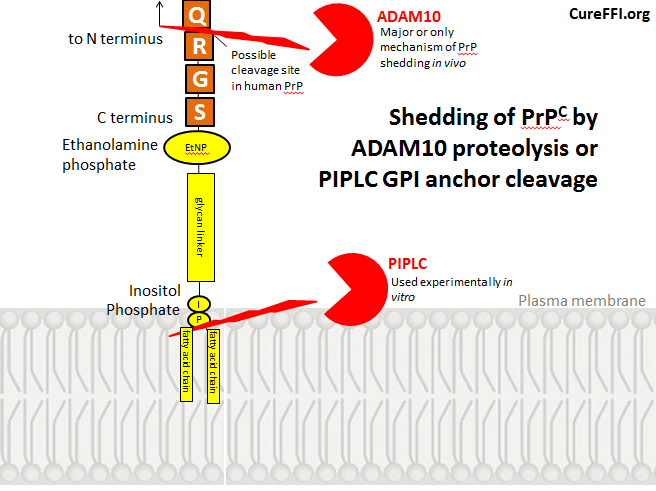
Shedding of PrPC was first explored in detail in cultured embryonic Syrian hamster neural tissue [Borcheldt 1993]. Cells were pulsed with 35S methionine and within 24 hours about 10 – 20% of the radiolabeled PrPC had been shed into the culture medium. This raised the question of where anchored PrPC was being cleaved – was the protein itself cleaved at a peptide bond between two amino acids, or was the GPI anchor being cleaved? To understand this question better, I had to get a grasp on GPI structure. Based on this, this, this and this, my best understanding of the GPI anchor is as follows:
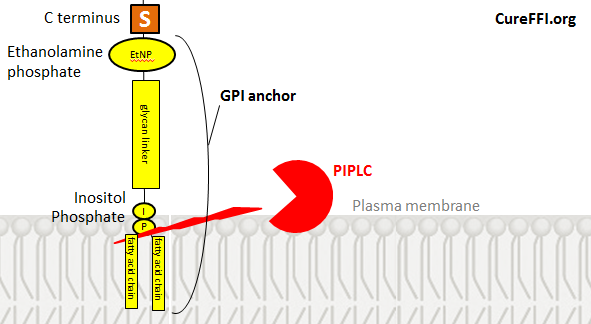
The C terminus of the protein is attached to ethanolamine phosphate is attached to a glycan is attached to inositol is attached to phosphate is attached to fatty acid chains. From inositol on downward it’s the same as PI, a component of the plasma membrane, so that part of the anchor stays embedded in the membrane just like any other phospholipid.
PI-specific phospholipase C (PIPLC), an enzyme which specifically cleaves GPI anchors, cleaves them between the phosphate and the fatty acid chains, thus leaving phosphate, inositol, the glycan chain and ethanolamine still attached to the C terminus of the shed protein. Hydrofluoric acid (HF) can remove these remaining portions of the GPI anchor following PIPLC cleavage. Therefore if you treat cells with PIPLC, run the shed PrPC on a gel, then treat the PrPC with HF and run it again, you’ll find that HF removes 2-4 kDa of PrPC’s molecular weight. That is your sign that the remnants of the GPI anchor were initially attached, but are no longer after HF treatment.
By contrast, Borcheldt found that the shed PrPC’s molecular weight was not affected by HF treatment, meaning the GPI anchor was already gone. Yet the shed PrPC still stained positive for the most C-terminal antibodies to PrP available at that time, suggesting most of the protein was intact. This meant that some proteolytic enzyme was cleaving PrP at a peptide bond very close to the C terminus. (People still use PIPLC as an experimental tool for releasing PrP from the cell surface but I haven’t read any reports claiming that GPI anchor cleavage is a mechanism for PrP shedding in vivo).
The exact site and mechanism of this proteolytic cleavage were later explored in more detail in HEK and N2a cells (human and mouse respectively) [Taylor 2009]. GPI-anchored PrP ends in QAYYQRGS in human, and QAYYDGRRS in mouse. The DG at this site appears fairly unique to rodents:

Note: I wasn’t happy with how T-Coffee aligned the C-terminus of mammal PrP so for this graphic I took the liberty of rearranging the way I think they line up best. I consider it more likely that the Q in most mammals’ fourth-to-last position corresponds to the G in rodents, rather than the D, since you can get between G and Q in one base pair change, while D to Q takes two changes.
Taylor found that the cleavage site in recombinant MoPrP is between the G and R (i.e. QAYYDG | RRS), a site that doesn’t exist in HuPrP. I couldn’t find any reports confirming the exact sheddase cleavage site is in HuPrP, but shedding is indeed observed in HuPrP just as in SHaPrP [Borcheldt 1993]. For the purposes of the diagram at the top of this post, I’ve drawn it as QAYYQ | RGS, but I haven’t actually read any evidence for this being the case.
Taylor also provided a variety of in vitro evidence implicating ADAM9 and ADAM10 as being responsible for this proteolytic cleavage (indirectly and directly, respectively) [Taylor 2009]. Of note, both of those are also believed to be involved in alpha cleavage of PrP – cleavage in the center of the protein, on either side of H111.
relation to bank voles?
How does ADAM10 “know” where to cleave PrP? Borcheldt speculated that PrP might be processed by “a molecule like the secretase that cleaves Alzheimer’s precursor protein (APP)… This secretase exhibits little amino acid specificity and appears to cleave APP at a defined distance from the membrane”. Two decades later, we know that the alpha-secretase that cleaves APP right above the membrane is none other than ADAM10 itself [Kuhn 2010] – the same sheddase that cleaves PrP. A review of known ADAM10 cleavage sites plus a battery of experiments with synthetic peptides [Caescu 2009, see esp. Table 2 and Figure 1] failed to find any obvious consensus sequence for ADAM10 cleavage – but amino acid sequence does appear to matter somewhat. Interestingly, if you look at the ADAM10 portion of the heat map in Figure 1, ADAM10 appears to ‘prefer’ E in the P2 position (two amino acids N-terminal of the cleavage site) over D.
This is of interest because bank voles are the only known mammal with an E in that position. Backstory: bank voles have almost no transmission barrier to infection by any prion strain [Nonno 2006, Agrimi 2008, Heisey 2009], and overexpressing wild-type bank vole PrP in mice causes a spontaneous transmissible prion disease [Watts 2012]. At Prion2013, Joel Watts presented his findings that mice expressing BvPrP, as it’s known, are highly susceptible to every prion strain tested. They have short to medium incubation times at first passage and very short (~35 day) incubation times on second passage. Weirdly, the BvPrP mice show almost no PrP-res, even when terminally ill of prion strains that normally produce PrP-res in other mammals. One interpretation of this is that BvPrP is especially prone to produce PrPL – the hypothesized [Hill 2000, Sandberg 2011] lethal by-product of prion conversion, and that therefore it kills BvPrP mice earlier on, before they’ve had a chance to accumulate as much PrP-res.
I had the chance to talk to Joel Watts later that day and I chatted him up about the differences between bank vole PrP and mouse PrP. He said they differ in 9 amino acids [see his mouse / bank vole alignment], but 8 of those changes have been seen in other mammals. The only sequence change that is (as far as we know) absolutely unique to bank voles is the E near the C terminus:
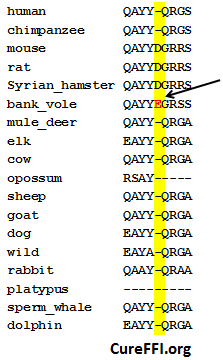
Watts opined that the exceptional instability of BvPrP could be due to this change, but that this would be surprising, since D→E is a fairly conservative amino acid change and since the location of this change is so C-terminal, far from the prion conversion action at the core region. Alternately, the unique properties of BvPrP could owe to bank voles’ particular combination of other amino acid changes.
The E could potentially matter if it influenced shedding. It’s not clear how much the E would really increase ADAM10′s affinity for the C terminus of PrP, though, considering that the overall sequence (EGRSS) still doesn’t appear to be highly favored by ADAM10 [Caescu 2009, Table 2 and Figure 1]. It’s also not clear how a hypothetical increase in shedding would affect prion disease progression. When Andy Hill presented at Prion2013, he cited three major mechanisms – exosomes, cell-to-cell contact, and tunneling nanotubes – that allow prion infection to spread from cell to cell. There was no mention of shed PrP as a mechanism for the spread of regular prion diseases – though anchorless PrP does cause a different disease, cerebral amyloid angiopathy – more on this below. Anyway, even if shedding does help prions spread more rapidly, that wouldn’t explain other strange features of BvPrP – notably, the relative absence of PrP-res.
therapeutic value of ADAM10?
One study created transgenic mice overexpressing ADAM10, and found that they had extended survival times after scrapie infection [Endres 2009]. The overexpressers lived to an average of 153 days, controls lived to 134 days – a 12% delay, p < .0001 according to the author. There are at least three possible ways that ADAM10 overexpression may have affected the course of disease in these mice. First, Endres found that PrP mRNA was reduced by about 30% in the ADAM10 overexpressers – the transgene had apparently suppressed transcription of Prnp. The mechanism for that is not at all clear. Second, we know ADAM10 is involved in alpha cleavage of PrP, which is believed to render the remaining C1 fragment incapable of converting to PrPSc, so that would be expected to slow down prion disease progression. Probably either of these first two explanations – mRNA reduction or alpha cleavage – could be sufficient to explain a 12% extension of survival time in Endres’s mice. Third, ADAM10 is involved in shedding of PrP [Taylor 2009], which might be good or bad for prion disease, as discussed in the next section.
therapeutic implications of shedding and anchorless PrP
What implication does shedding have for prion disease progression? Is there a potential therapeutic angle here? It’s not totally clear.
There is a school of thought that cell surface localization of PrP is essential for disease. For instance, the recent Scripps FRET assay [Karapetyan & Sferrazza 2013] was conceived with the goal of detecting compounds that reduce cell surface PrP (though in practice the design of the assay would not have detected compounds that increased shedding). The main argument for cell surface localization being crucial to disease is that when ScN2a (scrapie-infected mouse neuroblastoma) cells are treated with PIPLC for 3 days, they are “cured” of scrapie infection [Enari 2001]. There has been a bit of other work in this area as well – glimepiride, a drug that promotes GPI cleavage, can also reduce PrPSc formation in cell culture [Bate 2009].
But the kinetics of prion infection in rapidly dividing N2a cells are quite different than the kinetics of prion infection in post-mitotic neurons in vivo. Cell division constantly dilutes prions in ScN2a cells, and the sum of dilution plus degradation allows the cells to keep pace with prion formation, avoiding a toxic buildup of prions [Ghaemmaghami 2007]. In Enari’s experiment the cells were not split or passaged, meaning that cell division was somewhat limited and shed PrPSc was not washed away but rather stayed with the cells. That makes Enari’s result a bit more like in vivo conditions, but still, cell cultures aren’t animals, and they don’t have blood vessels where PrPSc plaques could build up.
In humans, premature stop codons resulting in truncated, anchorless PrP cause a form of cerebral amyloid angiopathy (CAA). This form of CAA is a prion disease characterized by intense amyloid deposition and an Alzheimer’s-like clinical presentation with a long disease duration (~ 4 years) and no spongiform change. This disease can result from Y145X [Kitamoto 1993, Ghetti 1996], Y163X [Revesz 2009], Y226X, Q227X [Jansen 2010] and probably also the recently reported frameshift mutation at codon 178 [Matsuzono 2013].
Based on the human data alone, it is impossible to disentangle whether anchorless PrP is inherently bad or if these mutations cause disease only because PrP is also truncated. The first mice expressing anchorless PrP were originally reported to have no spontaneous disease on their own, but to be capable of producing amyloid plaques upon prion infection, albeit without fatal disease and with a very different histopathological phenotype than prion-infected wild-type mice, similar to CAA [Chesebro 2005]. This was later revised when it was found that these mice do have behavioral deficits after prion infection [Trifilo 2008] and then revised again when it was discovered that homozygous anchorless PrP mice, when inoculated with prions, did indeed get a fatal disease dubbed “fatal transmissible amyloid encephalopathy” [Chesebro 2010].
The course of prion disease in Chesebro’s homozygous anchorless mice differs from wild-type mice in two ways: first, the phenotype is characterized by plaque deposition, much like the human CAA diseases, and second, the incubation time for prion inoculation is two or three times longer than for wild-type mice: 300 – 480 days instead of 150 days [Chesebro 2010]. That latter point sounded promising – if we could find drugs to promote PrP shedding, could we “convert” prion disease into CAA and thus slow the disease down by three-fold? At Prion2013 I asked Bruce Chesebro whether this longer incubation time was an inherent property of anchorless PrP or was simply due to reduced expression, as anchorless PrP tends to be degraded before reaching the cell surface – Chesebro’s mice express just ~0.1x wild-type levels of PrP, which could be sufficient to explain the long incubation times. From that particular mouse model, it’s impossible to tell.
But another anchorless mouse model expressing ~1x PrP is not only susceptible to prions but even develops a spontaneous prion disease eventually [Stohr 2011]. And the disease is even worse if you co-express anchored PrP with anchorless PrP. Thus it seems that promoting GPI anchor cleavage and/or proteolytic shedding of PrP in vivo might well do more harm than good.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.