Measuring mHTT levels in CSF in Huntington disease
In my last post I reviewed the completed clinical trials in prion disease, all of which have been conducted in symptomatic individuals and most of which have simply measured time to death as a clinical endpoint. If we ever want to be able to conduct clinical trials in asymptomatic individuals harboring PRNP mutations that cause genetic prion disease, then we’re going to need a biomarker that tells us whether the patients are getting closer to disease onset, and whether a drug is slowing or reversing that progression. Right now, we don’t have any such biomarker. There have been a few intriguing suggestions, such as the N=1 case study raising the possibility that FDG-PET may be able to detect subtle decreases in thalamic metabolic activity a year or two before onset of fatal familial insomnia [Cortelli 2006]. But we don’t yet have any prion biomarkers observable a larger number of years prior to disease onset, nor any biomarkers that have been validated in a large cohort. So if one day we do have a drug we want to give to asymptomatic people, how will we ever know if it is working?
Researchers in the Huntington disease field have the same problem. Indeed, because Huntington disease is much more slowly progressive, with survival times of perhaps 20 years [Foroud 1999] compared to just 5 months in prion disease [Pocchiari 2004], they need biomarkers even to study symptomatic patients.
This month, there is a pretty exciting, if still preliminary, finding in the Huntington disease field. Researchers were able to measure huntingtin levels in human cerebrospinal fluid, and showed that the level of mutant huntingtin (mHTT) is higher in symptomatic than presymptomatic mutation carriers, and is correlated with various measures of disease progression [Wild & Boggio 2015].
An overlapping group of researchers had previously managed to quantify mHTT in blood using a FRET assay, and showed that mHTT levels in blood correlated with disease progression [Weiss 2012]. You might ask, if we already had a biomarker in blood that’s correlated with disease progression, then why do we care about measuring mHTT in CSF? After all, CSF samples come in small batches (you can’t take more than about 10 mL at a time) and the procedure is more invasive than a blood draw. But Huntington disease is a disease of the brain, and treatments need to target the brain. We don’t really know of any blood phenotype in HD (though perhaps there is something subtle we haven’t noticed) and so we don’t really care about treating the blood. If we had a therapeutic that corrected some defect in blood and made it look like everything was fine from the blood perspective but didn’t actually get into the brain and make a difference there, then a blood biomarker would be misleading. Most importantly, antisense oligonucleotides (ASOs) to lower HTT levels are in the pipeline to begin phase 1 clinical trials later this year. The plan is to administer the drug by injection into the CSF via the intrathecal space at the base of the spine. The hope is that the ASOs will lower HTT levels in the brain by ~50%, comparable to what has been seen in animals [Kordasiewicz 2012], but we don’t know how much (if at all) they will lower HTT levels in peripheral tissues, so again, what we want to measure is what’s going on in the brain, and CSF is the closest proxy we can get.
Huntingtin is particularly hard to measure in CSF because the protein is intracellular, localized mostly in the cytosol. The FRET assay they had originally used in blood [Weiss 2012] only works because you can collect circulating white blood cells, lyse them open with detergents, and then measure the protein that was inside. But that trick doesn’t work with CSF, because normally CSF contains almost zero cells - it’s just a solution of protein, glucose and ions. So for this study, they used a highly sensitive Singulex single molecule counting assay to quantify the rather minute (femtomolar) concentrations of mHTT that are floating around in the CSF. This process involves first using capture antibody specific to HTT, and then using a detection antibody specific to only mHTT, as it reacts with the expanded polyQ tract that causes the disease.
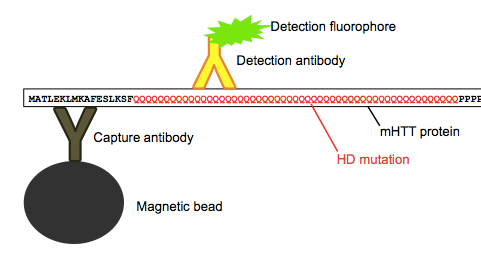
Above: as far as I can tell, this is how the assay works.
They found that the mHTT levels they measured in CSF were correlated with levels of two other neuronal proteins, tau and neurofilament light chain (NFL), which suggests that the mHTT they measured in the CSF came from neurons - perhaps following neuronal death or injury - and not from blood contamination or some other source. mHTT was detected in all but two people with the HD mutation (more on those two in a minute), and there were no false positives from any control individuals who lacked the HD mutation. Among people with the HD mutation, the level of mHTT detected in CSF was, on average, higher in symptomatic patients than in presymptomatic carriers. What’s more, the level correlated with a couple of different measures of HD progression. One such metric, known as the “disease burden score”, is simply a person’s CAG repeat length minus 35.5, times their age [Penney 1997]. Obviously, this incredibly simple statistic is not measuring anything about the disease progression of the individual, but rather simply reflects the population average of how the disease progresses. Nonetheless, it was decently correlated with mHTT levels, with R = 0.7. I noted with interest that the two asymptomatic mutation carriers in whom zero mHTT was detected had two of the lowest disease burden scores of anyone (see two dots in lower left of Fig 3B - they either have very low CAG, or are very young, so the failure to detect mHTT in these individuals may not be an assay failure, but rather may well mean that these people simply aren’t shedding any mHTT into their CSF. Finally, and perhaps most compellingly, mHTT levels were also significantly correlated with the Unified Huntington Disease Rating Scale (UHDRS) motor score, which actually is a measurement of the individual’s phenotype, even after controlling for disease burden score.
These results raise the exciting possibility that mHTT levels in CSF could function as a biomarker in clinical trials. This is particularly relevant to the ASO therapy headed for phase 1 trials soon. The ability to measure mHTT in CSF means we’ll be able to tell whether ASOs are successful at reducing HTT levels.
To be sure, it’s still early days. This study showed that mHTT levels are correlated with clinical severity cross-sectionally (across different patients), which leads us to the compelling - but still untested - hypothesis that mHTT levels may also be correlated with clinical severity longitudinally. Testing that hypothesis will require regular CSF collections from patients followed prospectively, an effort that I’ll wager the authors of this paper have already begun, but which will take years to complete. It also remains to be seen whether the assay will be sufficiently precise and reproducible to detect knockdown of mHTT in individual people. Note that in the current study, symptomatic patients had higher mHTT levels than presymptomatic people on average, but not categorically. The assay may also prove sufficiently precise to discriminate a treated group of patients from an untreated group of patients, but we can’t yet take that for granted - only experimentation will tell. Finally, the biggest question, which only clinical trials will be able to answer, is whether changes in mHTT levels due to drug treatment will be predictive of altered clinical outcomes in patients.
That’s an incredibly important question, and the possibility that the answer could be “yes” is one reason why this study is so exciting despite all the unknowns. As an analogy, consider mipomersen, currently the only FDA-approved ASO drug. This drug is approved for people with a particular form of familial hypercholesterolemia, and it acts by knocking down expression of the APOB gene, which encodes a component of low density lipoprotein (LDL or “bad cholesterol”) particles. In the clinical trial that led to mipomersen’s approval, it was demonstrated that LDL cholesterol dropped 24.7% in people treated with mipomersen for six months, compared to 3.3% in people treated with placebo [Raal 2010]. If it hadn’t been possible to measure LDL cholesterol as a biomarker in that trial, the only way to figure out if the drug was working would be to wait years and years to see if people in the placebo group had a statistically higher rate of heart attacks. Similarly, in HD trials, no one wants to wait years (decades!) to see when people die of HD. Whether or not FDA would ever be willing to accept mHTT or any other biochemical (as opposed to clinical) biomarker as the basis for approving an HD drug remains to be seen, but regardless, biomarkers can provide critical early results that influence trial design (for instance, identifying patient subgroups that respond well) and sustaining the enthusiasm of patients deciding whether to enroll, and drug companies deciding how much more to invest.
The success so far with measuring mHTT in Huntington disease makes me wonder about the possibility of measuring PrP levels in CSF as a biomarker for prion disease. My next post will be a roundup of the literature on PrP levels in the CSF, and an evaluation of whether this has any merit as a potential marker of prion disease progression.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.