Polythiophenes go in vivo
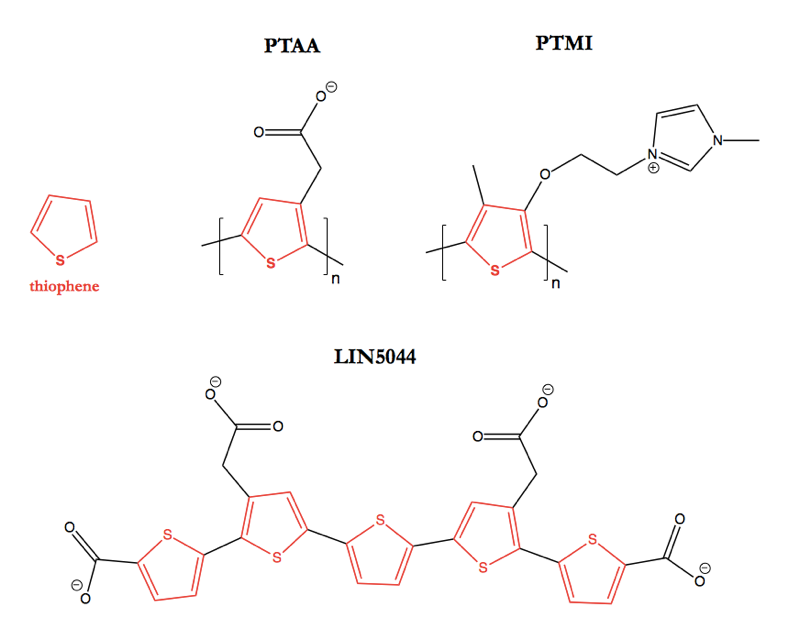
Above: The structures of some polythiophene compounds discussed in this post. Highlighted in red is the eponymous thiophene group.
Polythiophenes — chains of several thiophenes strung together at the 2 and 5 positions — are probably better known to material scientists than to biologists. Molecules of this class have been studied as potential materials for making chemical sensors [Ho & Leclerc 2003, Wang 2008], actuators [Smela 2003], semiconductors [Heeney 2005, Clark 2007, Kane-Maguire & Wallace 2010], and solar cells [Kim 2006].
Some of the same qualities that make polythiophenes interesting to material scientists make them nuisance compounds from the perspective of chemical biologists. Polythiophenes are optically active and often form aggregates in poor solvents [Langeveld-Voss 2000, Hoeben 2005, Goto 2012, Spano & Silva 2014]. Photons are usually the basis of assay readouts, and aggregation is well-known as a source of non-specific inhibition [McGovern 2002], so optical activity and aggregation create opportunities for false positives in chemical library screens. One individual I spoke to even labeled polythiophenes as pan-assay interference compounds or “PAINS” [Baell & Walters 2014].
Perhaps for these reasons, some people with whom I spoke reacted with initial disbelief last month when the Aguzzi lab reported that a series of polythiophenes possess antiprion activity in vivo [Herrmann 2015]. Surely this was just a false positive screening hit, and surely there were the usual reasons to be skeptical of the in vivo data (see e.g. [Ioannidis 2005, Scott 2008])? But this is real. The study is thorough, some of the effect sizes are quite large, and polythiophenes are not even a high throughput screening hit at all — in fact, the new study is but the latest chapter in a decade-long series of investigations into the ability of certain polythiophenes to bind amyloids, discriminate different protein conformations, and inhibit prion propagation. In this post I will review some of this history and then discuss the prospects for polythiophenes as therapeutics.
In the literature on polythiophenes as anti-amyloid agents, the compounds are sometimes referred to as conjugated polyelectrolytes or luminescent conjugated polymers (LCPs). The new study is the first to focus largely on well-defined small molecule structures; most of the earlier studies used polythiophene acetic acid (PTAA), polythiophene methyl imidazole (PTMI), or other polymers of heterogeneous length. The interest in polythiophenes as probes for amyloid diseases began with studies of PTAA as a probe for imaging amyloids made of bovine insulin or chicken lysozyme [Herland 2005, Nilsson 2005]. It was found that PTAA’s peak absorbance and emisson wavelengths both undergo a red shift upon exposure to amyloids, apparently due to changes in the conformation of the polythiophene backbone. This meant that a ratio, such as the ratio of PTAA’s emission at 550 nm (its peak in the absence of amyloid) to 580 nm (its peak in the presence of amyloid) could be used to monitor the kinetics of amyloid formation in vitro, or to discriminate an amyloid from a non-amyloid protein fibril such as collagen.
Those were all in vitro experiments using purified protein, though, so there were not yet any data on how specific of a stain PTAA would prove to be — if confronted with the myriad proteins of a cell or whole tissue, would it still single out the amyloid? The next year, the same group was back with additional data showing that PTAA could indeed be used to stain amyloid plaques in whole tissue sections — insulin amyloid in human pancreas, Aβ in human brain, and light chain amyloids in several tissues [Nilsson 2006]. Although this was not set up as a diagnostic study using a series of cases and controls to quantify the sensitivity and specificity of the signal, the selected images in the paper at least showed that PTAA was binding to specific deposits and not just sticking wholesale to every molecule in the cell.
Peter Nilsson, who had pioneered much of the PTAA work, soon moved to Zurich and began working with Christina Sigurdson and Adriano Aguzzi on applying polythiophenes to the study of prions in histological sections. They found that not only did PTAA and PTMI stain prion-infected brains and not uninfected brains, they also exhibited staining properties specific to different prion strains [Sigurdson & Nilsson 2007]. To rule out the confounder of different primary structures of PrP, all of the experiments were performed with various prion strains passaged into Tga20 mice, which express ~10x wild-type levels of wild-type MoPrP-A [Fischer 1996]. PTAA stained mouse-passaged CWD and sheep scrapie isolates but not RML or mouse-passaged BSE prions. PTMI stained CWD and RML but not BSE or scrapie. What’s more, the emission spectra were different for different prion strains — the emission spectra for PTAA bound to CWD peaked more sharply at 532 nm than those for BSE or scrapie, and ratios of different wavelengths could be used to discriminate these three prion strains and even different serial passages of CWD. These emission spectra reflected different conformations of the polythiophene backbone, and thus were taken to imply different conformations of the PrP plaques to which PTAA was bound. Thus, this study became yet another line of evidence that distinct prion strain properties are encoded in different protein conformations. Over the next few years, polythiophenes were similarly applied to discriminating different strains of PrP prions and of various other amyloids [Aslund 2009, Nilsson 2010, Sigurdson 2010, Klingstedt 2011].
At the same time, the Aguzzi lab hypothesized that polythiophenes might possess activity as antiprion compounds. After all, over the years, several other compounds that were initially studied as probes for amyloid imaging have also turned out to inhibit prion propagation by one metric or another. For instance, Congo red, the tautological amyloid stain, also reduces the accumulation of proteinase K-resistant PrP in infected cell lines [Caughey & Race 1992]. (Note that although Congo red is a notorious aggregator and promiscuous binder [McGovern 2002], the readouts for those experiments were Western blots; this wasn’t simply a false positive hit in a high-throughput screen). And cpd-b, the first orally administered drug ever to substantially delay the onset of disease after intracerebral prion infection [Kawasaki 2007], grew out of a search for amyloid PET ligands [Okamura 2004, Ishikawa 2006].
And indeed, it turned out that PTAA did indeed inhibit prion replication in a slice culture model [Margalith 2012]. The Aguzzi lab uses cerebellar organotypic slice cultures, which are 350 μm thick sections of neonate mouse cerebellum, as a model of prion pathogenesis capable of recapitulating both prion replication and neurotoxicity [Falsig 2008, Falsig & Aguzzi 2008, Falsig 2012]. In these slices, treatment with 10 μg/mL of PTAA from early timepoints reduced PK-resistant PrPSc accumulation to nearly undetectable levels (see Figure 5). The amount of PTAA is expressed in wt/vol because the PTAA used here is a polymer of variable length. The monomeric unit has a molecular weight of 169.22 Da, so the equivalent monomer concentration here works out to .01 g/L / 169.22 g/mol = 59 μM.
The new paper out last month [Herrmann 2015] extends those ex vivo results in vivo through an iterative series of survival studies in RML-infected mice along with in silico studies of mechanism and synthesis of new, more effective polythiophenes. The study begins with an “LCP mix” including a mixture of PTAA polymers, infused directly into the mouse brain via intraventricular Alzet pumps. The initial hints of efficacy from that in vivo trial lead to the synthesis of several new polythiophenes with specifically defined structures (as opposed to being random polymers), one of which was even tested via intraperitoneal injection rather than intraventricular infusion. Several polythiophenes prove effective at delaying prion disease in these mice, of which one dubbed LIN5044 (top) is the most effective. Here are the survival data for that compound. These are all in PrP-overexpressing mice, so 49 dpi represents a timepoint about ¾ of the way through the disease course, right around the onset of earliest symptoms:
| prion strain | timepoint | drug administration route | delay in disease onset | p value |
|---|---|---|---|---|
| RML | prophylactic | intraventricular | +87% | .001 |
| RML | 49 dpi | intraventricular | +24% | .002 |
| RML | 49 dpi | intraperitoneal | +12% | .04 |
| 263K | 20 dpi | intraperitoneal | +17% | .0003 |
To see how these results stack up against those of other antiprion compounds, I made the below timepoint-delay plot of LIN5044 versus anle138b, cpd-b, and IND24. The x axis is the time in the disease course when treatment began, and the y axis indicates by what fold survival time was increased.

Data, code to produce above plot.
The results for the later, symptomatic, timepoints are comparable to results reported for IND24 and cpd-b in transgenic PrP overexpressers [Kawasaki 2007, Giles 2015]. Earlier in the disease course, LIN5044 is clearly less effective than the other compounds.
Meanwhile, most of the experiments in this paper were performed using direct intraventricular infusion, “to attain adequate local doses and circumvent brain penetrance issues” [Herrmann 2015], whereas IND24, anle138b, and cpd-b have all been shown effective when administered orally. The paper does not include any explicit measurements of pharmacokinetic parameters, so the only evidence that LIN5044 is capable of crossing the blood-brain barrier is the fact that it was effective when adminstered intraperitoneally in the latter two experiments of the table above. It would be of interest to know whether limited drug availability in the brain is one reason why the effect sizes in those two experiments are quite small. One medicinal chemist with whom I spoke asked me whether they had used enormous doses to force some of the drug across the BBB, but for what it’s worth, the doses used in this paper are actually pretty low. For the survival studies with intraperitoneal drug delivery, the dose was 400 μg three times a week. Assuming, say, a 30g mouse, that’s an equivalent of only about .4 mg / .030 kg * 3/7 = 6 mg/kg/day. That’s an order of magnitude lower than the doses used for IND24 (50 to 210 mg/kg/day) [Giles 2015]. The intraventricular infusion experiments used .15 μL/hour of a 4 mg/mL solution, which works out to only about 0.5 mg/kg/day, though of course the numbers are not directly comparable where direct infusion into the brain is concerned. If LIN5044 does indeed cross the BBB, that is somewhat surprising. In terms of its size and hydrophilicity, LIN5044 falls slightly outside the space of CNS drugs:

Code to produce above plot.
And what this plot doesn’t even show is LIN5044’s net charge of -4. As shown here, virtually all FDA-approved CNS drugs are neutral, and none have an absolute charge greater than ±2. And LIN5044’s total polar surface area (tPSA) is 160.52 Å2, higher than any approved CNS drug (also discussed here).
No doubt, the experiments here are presented as a proof of principle, and LIN5044 is presented as a lead, not a drug. So perhaps it’s not fair to compare its chemical properties to those of FDA-approved drugs. Yet the data in the paper do not inspire great optimism about the prospects for medicinal chemistry optimization of polythiophenes. Based on the exploration of the polythiophenes’ structure-activity relationships throughout the paper, using in silico models, binding measurements to recombinant PrP fibrils, and the in vivo data, the Discussion concludes that for antiprion activity it is essential for a polythiophene to have a backbone of at least 5 thiophenes, and to have negatively charged side groups. Based on these constraints, it would seem rather challenging to make LIN5044 any smaller, less charged, or more hydrophobic while preserving its antiprion activity. And medicinal chemists with whom I spoke were pessimistic about the prospects for optimization of these compounds.
Perhaps the medicinal chemistry would be a challenge worth struggling to overcome if the polythiophenes actually worked against human prions, which IND24, cpd-b, and anle138b have all failed to do. To address the issue of prion strain specificity, which has been the Achilles heel of those other antiprion leads, the authors performed one experiment in which LIN5044 was tested against 263K prions in mice expressing hamster PrP. The compound was found to be effective, increasing survival time by +17%. So LIN5044 works against at least two prion strains. But that’s also true of IND24, which works against both mouse RML and ME7 prions as well as two different CWD isolates, from elk and white-tailed deeer [Berry 2013, Berry 2015]. In fact, cpd-b was (albeit only marginally) effective against 263K prions [Kawasaki 2007], yet still had no effect against MM1 sCJD prions [Lu & Giles 2013]. So the only way to know if polythiophenes would be effective against human prions would be to test them in mice infected with human prions, which hasn’t been done yet.
Absent any data regarding effects on human prions, and considering their troublesome SAR and lesser efficacy compared to other antiprion leads, I am not optimistic about the prospects for polythiophenes to ever become drugs. The new study is still interesting, though, particularly as it sets the stage for some discussions about antiprion mechanisms of action and PrP structural biology.
The new paper and earlier work from the Aguzzi lab present a series of lines of evidence to suggest that the compounds bind to the amyloid backbone and stabilize PrPSc [Margalith 2012, Herrmann 2015]. The notion that stabilizing prions could be a therapeutic strategy seems counter-intuitive at first. To make sense of it, it is useful to return to the “breakable filament” model of prion propagation [see e.g. Knowles 2009]. This model holds that prions are fibrils which can grow linearly by add monomers at both ends, but which must break in order to form new ends and achieve exponential growth:
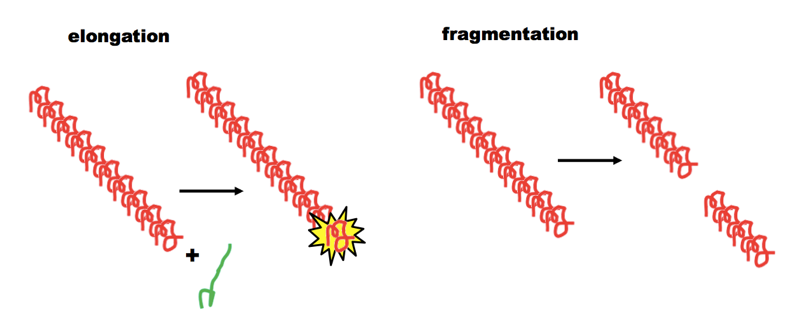
Thus, if you can keep the fibrils from breaking, you can limit them to linear growth. In support of this idea, there is evidence that less frangible (less breakable) yeast prions have “weaker” phenotypes and are more likely to self-cure — see this post and [Tanaka 2006].
Do the polythiophenes achieve their antiprion effects by inhibiting fragmentation? It is worth going through the evidence step-by-step. First, it seems very likely that the effect is indeed mediated by direct binding to PrPSc rather than some other target. Otherwise, it would be impossible to explain the selective staining of amyloids in histological sections and in vitro studies, or the differential optical properties of polythiophenes upon binding to different prion strains. The new paper also suggests a specific binding mode on the basis of a series of in silico models and in vitro experiments plus the in vivo data. This begins with in silico docking of a polythiophene onto the backbone of HET-s, the only prion and the only amyloid for which a high-resolution structure has been solved [PDB# 2RNM]. HET-s is a β-solenoid with mixed intra- and inter-molecular β-sheets (each molecule occupies two rungs of the coil). The docking studies suggested that polythiophenes could fit into a groove on the HET-s backbone, and that each anionic group on the polythiophene would bind to a positively charged lysine on the backbone. Next, they considered a model structure of fibrils containing a PrP repeat sequence and modeled the docking of polythiophenes onto that backbone as well. Subsequent empirical data for actual fibrils made of recombinant mouse PrP 23-231 showed that the polythiophenes predicted to bind most strongly in silico also had the lowest KD (strongest affinity) in vitro, and also the greatest activity in vivo. This all seems pretty plausible, but I am not enough of a structural biologist to really evaluate the claims. I do suspect that these results will not be enough to convince everyone that this is indeed the exact binding mode of the polythiophenes, since the debate over whether PrPSc is a β-solenoid or a parallel in-register intermolecular β-sheet (PIRIBS) structure continues to run strong.
Regardless of whether polythiophenes do bind to the amyloid backbone in the specified fashion, there is also the question of whether their effect is indeed to stabilize fibrils and reduce frangibility. Alternative hypotheses might be that they inhibit the elongation of fibrils, or destabilize the fibrils, rendering them more prone to degradation. The original evidence for the idea that polythiophenes stabilize fibrils comes from a series of experiments reported a few years ago [Margalith 2012]. They investigated how several polythiophene compounds affected the properties of RML PrPSc in a series of assays. Here’s a breakdown of some of the results:
- They reported that co-incubation of polythiophenes with RML brain homogenate increased the resistance of PrPSc to proteinase K digestion, although when I look at Figure 1, the dose-response is not visually obvious to me for any polythiophene except one (pHTAA).
- In the scrapie cell endpoint assay — a measure of what proportion of cells become infected and produce PK-resistant PrPSc in response to prion exposure [Klohn 2003] — the polythiophenes had a more clear dose-dependent effect, of reducing the ability of infected brain homogenates to infect cells. It seems to me that one might expect this result for any antiprion compound, whether it inhibited elongation or inhibited fragmentation.
- Then there is the misfolded protein assay (MPA), which I had never understood until reading up on it now. Several years ago, David Peretz and colleagues screened a battery of PrP peptides and found that PrP23-30 and PrP100-111 both have the property of selectively binding PrPSc [Lau 2007]. This is biologically interesting in itself and cannot possibly be a coincidence, but it has also proven practically useful: they used bead-bound peptides to pull down PrPSc from blood with remarkable specificity despite the presence of plenty of PrPC and other proteins. The Aguzzi lab later adapted this assay, using a bead-conjugated peptoid called “PSR1” (which I believe refers to PrP23-30) to pull down PrPSc from brain homogenates, with subsequent immunodetection to quantify it [Margalith 2012]. Polythiophenes reduced the amount of PrPSc detected by this assay. The explanation for this is not clear to me, as prions do not have an opportunity to replicate in the course of this assay, so the amount of PrPSc should not change. Possibly polythiophenes could occlude the binding site of the peptides on the surface of PrPSc, but this is just speculation as we do not know the details of how these peptides bind PrPSc.
- Polythiophenes were found to increase the lability of PK-resistant PrPSc to guanidine denaturation, causing a loss of PK resistance at as little as 2M GdnHCl, compared to 3M for untreated samples. It seems to me that this result argues that polythiophenes de-stabilize, rather than stabilize, PrPSc fibrils.
- Polythiophenes were shown to alter the in vitro fibrillization of recombinant PrP, assessed by electron microscopy. Untreated PrP formed long, elegant fibrils. Treatment with high concentrations of polythiophenes completely abolished fibrils, while moderate concentrations yielded smaller, less ordered fibrils (Figure 6). This is the result I would expect if polythiophenes de-stabilized the fibrils and/or inhibited the addition of new monomers, rather than inhibiting fragmentation.
- In the new study [Herrmann 2015], by density gradient ultracentrifugation, the brains of mice treated with polythiophenes were shown to contain a different distribution of PrPSc aggregate sizes, with more small oligomers compared to large fibrils. As with the above bullet point, to me this seems more consistent with the idea that the compounds de-stabilize the fibrils.
Based on all of the above considerations, it seems clear that polythiophenes do bind to PrPSc, but I am not yet convinced that they act by stabilizing fibrils and reducing their frangibility, although it is an interesting hypothesis and I remain open to it. I welcome any comments from readers.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.