Food and Drug Law 06: Regulation of Drugs
These are my notes from class meeting 6 of Harvard Law School’s Food and Drug Law course, led by Prof. Peter Barton Hutt on January 10, 2017. Reading for today’s class meeting is pp. 77-78, 89-101, 641-751 and 957-990 of Food and Drug Law 4th Ed..
Definition of drugs
Drugs (and devices and biologicals) require premarket approval from FDA for safety and efficacy, whereas food and cosmetics are less tightly regulated, and FDA has little control over vitamins and supplements. This means the regulatory hurdle a product must clear depends heavily upon whether it can be classified as a “drug”, and therefore upon the definition of drug.
The FD&C Act section 201(g)(1) defines a drug as:
A) articles recognized in the official United States Pharmacopoeia, official Homeopathic Pharmacopoeia of the United States, or official National Formulary, or any supplement to them; and
B) articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals; and
C) articles (other than food) intended to affect the structure or any function of the body of man or other animals; and
D) articles intended for use as a component of any article specified in clause (A), (B), or (C)…
The Act’s definition of device has criteria exactly paralleling (A), (B), and (C) above, but applying to “an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part, or accessory… which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of its primary intended purposes.”
The courts have greatly limited the use of criterion (A) because it is based on inclusion in specific lists produced by private, non-governmental bodies. The United States Pharmacopoeia and National Formulary (USP-NF) that the FD&C refers to is a paywalled collection of “monographs” (see example) listing various standards and protocols for each drug. It’s produced by a non-governmental, non-profit organization called the United States Pharmacopeial Convention, better known as USP. USP’s list includes vitamins and minerals, and the Homeopathic Pharmacopeia also mentioned in (A) includes herbal supplements, yet none of these are now considered drugs by FDA, due to a series of court decisions. Most notably, in United States vs. Ova II (1975), the 3rd Circuit Court rejected FDA’s ability to regulate a pregnancy test kit included in USP, because even though the FD&C Act explicitly says that inclusion in USP is sufficient to qualify for FDA regulation, this “run[s] afoul of the principle that a legislative body may not lawfully delegate its functions to a private citizen or organization.” Instead, the court allowed a much more limited interpretation, that if the packaging is labeled “USP” to imply intended use as a drug, then FDA may enforce the product’s adherence to USP’s specified standards of quality and purity and so forth. If a substance is listed in USP but the packaging is not labeled USP, then FDA may not claim jurisdiction based solely on criterion (A). Obviously many things listed in USP do meet criteria (B) or (C) and thus can be regulated by FDA.
Criteria (B) and (C) both concern “intended use.” FDA’s own interpretation of “intended use” is that it refers to the intent on the part of “the persons legally responsible for the labeling of drugs,” and that this intent may be established by “labeling claims, advertising matter, or oral or written statements” or by the “circumstances surrounding the distribution.” These “circumstances,” such as the actions of downstream distributors, may in some cases suggest that the original manufacturer of the drug intended a use other than the use for which the product was explicitly labeled or advertised.
In practice, FDA almost always relies on statements explicitly made by the manufacturer or distributor who it’s going after. Only once has a court found evidence of “intent” absent any explicit statement: in United States v. Travia (2001), the courts found that selling nitrous oxide-filled ballons in the parking lot outside a concert, even absent any labeling or explicit oral or written statement, constituted sufficiently clear “intent” to allow FDA to intervene. Courts have also not been keen on allowing FDA to intervene based on the intent of a consumer or a third party (as opposed to manufacturer or distributor). In National Nutritional Foods Ass’n v. Mathews (1977), FDA had tried to regulate very high doses of vitamin A and D (>10,000 and >400 IU/day respectively) because naturopathic author Adelle Davis (not a vitamin manufacturer herself) had convinced readers to take huge doses and there were reports of toxicity. The courts decided that the dose cutoffs chosen were based on FDA’s concern about toxicity, not “intent.” (Note: that case concerned actions FDA had taken in 1972. By the time the case was decided, Congress had passed the Vitamin-Mineral Amendments of 1976, which thereafter explicitly forbade FDA from “classify[ing] any natural or synthetic vitamin or mineral (or combination thereof) as a drug solely because It exceeds the level of potency which [FDA] determines is nutritionally rational or useful”.)
Criterion (C) includes things like botulinum toxin, which is not intended to treat or prevent disease, but is intended to affect the structure or function of the body. As a historical matter, note that the 1906 Act only concerned treatment or prevention of disease; the inclusion of Criterion (C) was new in 1938. Criterion (C) explicitly excludes foods, but criterion (B) does not, and courts have therefore allowed FDA to classify some things as both drug and food. Alternatively, some things can be a drug in some cases and a food in other cases: FDA considers caffeine as a drug when it is the active ingredient in a pill but not when it is in a beverage.
The distinction between biologicals and drugs was re-defined on June 30, 2003 (see pp. 1133-1134). Monoclonal antibodies and other proteins (everything from interferon to insulin) are now evaluated by CDER, while CBER retains authority over vaccines, allergenic extracts, blood, antitoxins, cellular therapies, and gene therapies.
The distinction between drugs and cosmetics is a very fine line. Something marketed to “promote smooth, luxurious skin” would be a cosmetic, something marketed to “reduce the appearance of wrinkles” would also be a cosmetic, but something marketed to “reduce wrinkles” would be a drug.
Prescription vs. over-the-counter drugs
There have always been two categories of drugs — prescription and over-the-counter — but they were not originally codified in legislation. Manufacturers simply made a marketing decision, of whether they wanted to sell a drug directly to consumers, or sell it to pharmacists and require patients to have a doctor’s prescription. For years leading up to 1938, Congress unanimously agreed that manufacturers should make this decision and FDA should not have jurisdiction; in fact, this wasn’t even considered an issue for debate. Yet one year after the FD&C was enacted, FDA chose to establish a mandatory prescription category, arguing that there are some drugs for which labeling is not adequate to ensure patients will use it correctly. Pharmacists and doctors welcomed the new regulation, because it cemented their position, making it impossible for consumers to bypass them. Consumers, meanwhile, didn’t seem to notice the change. Congress eventually supported FDA’s decision by statute: the 1951 Durham-Humphrey Amendments added FD&C section 503(b)(1), establishing that certain drugs are “not safe for use except under the supervision of a practitioner licensed by law to administer such drug,” and therefore can only be sold per a prescription.
FDA usually approves new drugs as prescription-only, and will not consider over-the-counter (OTC) status until at least 5 years later. The rationale is that there could be adverse events that turn up only rarely or over the long term, and you need 5 years of postmarketing data to be sure enough of the risk-benefit tradeoff to allow consumers to directly purchase the drug. FDA can make the prescription-to-OTC switch of its own accord or in response to a drug manufactuer’s petition (for on-patent drugs) or in response to anyone’s petition (for off-patent drugs). FDA usually requires a study showing that consumers can understand the label, and a “home use study” showing that consumers actually follow the label. Drug companies are often eager to obtain OTC status. The 10 best-selling OTC drugs today were all prescription drugs 20 years ago (citation needed).
In the U.S., once OTC status is awarded, manufacturers can choose whether they want their product to be over-the-counter or “behind-the-counter”, meaning you don’t need a prescription but you do need to approach the counter and interact with the pharmacist on duty before purchasing. This is not a legal category but merely a marketing decision: companies believe that consumers may perceive behind-the-counter drugs as more safe and effective, and therefore be willing to pay twice as much. There exists no federal, statutory, behind-the-counter status in the U.S. The one exception is pseudoephedrine, which became effectively behind-the-counter with the Combat Methamphetamine Epidemic Act of 2005, but this is technically governed by Drug Enforcement Agency (DEA) regulations and does not formally establish a behind-the-counter status in FDA regulations. Some proposed establishing a behind-the-counter status for Plan B emergency contraception, and it has been proposed again recently for birth control, but none of these proposals have taken effect.
FDA rejected OTC status for statins [Strom 2005]. That article argues that most drugs switched from prescription to OTC status are drugs where it is easy for the consumer to self-diagnose the condition and to detect the therapeutic effect of the drug. In contrast, you can’t feel what your LDL cholesterol level is, nor can you feel whether a statin has lowered it. In addition, while statins are pretty safe for most people, they are contraindicated in certain liver conditions (which are not self-diagnosable) and may be unsafe in pregnancy, and compliance with treatment regimens is not ideal. In the U.K. there does exist a statutory behind-the-counter status, and some statins have this status.
Over-the-counter status for oral contraceptives continues to be a subject of debate. President-elect Trump endorsed OTC status last year, three states (CA, OR, WA) have enacted state legislation making birth control available behind-the-counter, and some members of Congress have pushed for birth control behind-the-counter status nationally; the main debate seems to be over worries about health insurance coverage if birth control becomes OTC. Although birth control is still prescription-only, FDA did approve OTC status for the Plan B emergency contraceptive after a legal effort by a public interest group, see Tummino v. Hamburg (2013).
An FDA study of birth control found that 80% of women using it for the first time actually do read the patient package insert with directions on how to take it. Excited about this result, FDA briefly required that all drugs have a patient package insert, but this occurred at the very end of President Carter’s term and was almost immediately revoked by the Reagan administration. FDA later decided to selectively require patient package inserts only for selected products. FDA also later established the now-familiar “Drug Facts” label standard for OTC drugs, in 1997 (62 FR 9024).
The experience with package inserts made FDA start to think that direct communication between drug companies and consumers could be a good thing, and so it allowed advertising, but initially with severe restrictions on the content and even the hour of the day when the ads could air on TV. Pharmaceutical companies threatened a First Amendment lawsuit, and FDA backed down and allowed more flexible advertising at all hours of day, which continues to present day. There is ongoing debate about whether these ads can make claims other than what is explicitly approved in the drug’s labeling, such as promoting off-label uses. FDA has attempted to crack down on certain advertising claims, but has lost in the lower courts and has decided not to appeal its losses to the Supreme Court, for fear that the Supreme Court will decide that the First Amendment broadly protects drug makers’ right to make any claim they want in ads. Note that for many drugs, a particular off-label use has become standard of care, and yet this use remains off-label, because if the drug is off-patent, no one has an incentive to pay for the clinical trial that would add this use to the labeling. Current Supreme Court precedent does hold that “mislabeling” in ads is not protected by the First Amendment, and so to steer clear of potential warning letters, many drug makers will clear their ads with FDA before running them.
Historical perspective on regulation of efficacy
In lecture 1 it was explained that FDA did not begin requiring premarket review of drug efficacy until the 1962 Drug Amendments. But there has been at least some regulation of efficacy going back decades earlier.
There are precedents even prior to 1906. During the Mexican-American War (1846-1848) the U.S. purchased drugs for its troops and found a stunning lack of quality control and prevalence of contamination in the commercial drug supply. The Import Drug Act of 1848 therefore authorized the Treasury to evaluate drugs to determine whether they were suitable for the government to purchase. This remained in effect until 1926, though Prof. Hutt hasn’t found much evidence that it was really used. The Biologics Act of 1902, described in lecture 1, also provided for evaluation of efficacy of biologicals, particularly vaccines.
When the federal government first began regulating drugs through the 1906 Food and Drugs Act, it focused mostly on safety. It did outlaw “misbranding”, but this was taken to concern only the ingredients or purity, rather than efficacy claims. In fact, the prevailing view at the time was that the regulation of efficacy would be impossible. A recent Supreme Court decision, School of Magnetic Healing v. McAnnulty (1902) had more or less decided as much. The American Judicial School of Magnetic Healing was an institution that taught mind-over-body healing, on the belief that “the human race does possess the innate power, through proper exercise of the faculty of the brain and mind, to largely control and remedy the ills that humanity is heir to”. The Postmaster General deemed their business to be a form of mail fraud and had ordered mail addressed to the school to be seized and returned to sender. The Supreme Court found the Postmaster to be in the wrong, deciding that:
Just exactly to what extent the mental condition affects the body, no one can accurately and definitely say… There is no exact standard of absolute truth by which to prove the assertion false and a fraud. We may not believe in the efficacy of the treatment to the extent claimed by complainants, and we may have no sympathy with them in such claims, and yet their effectiveness is but matter of opinion in any court… Unless the question may be reduced to one of fact as distinguished from mere opinion, we think these statutes cannot be invoked for the purpose of stopping the delivery of mail matter.
So under the original 1906 law, when FDA did at one point attempt to enforce against a false claim of anti-cancer efficacy, the courts threw it out in United States v. Johnson (1911), citing the above decision. Congress responded by passing the Sherley Amendment (1912) which made it so that claims of “curative or therapeutic effect” that are both “false and fraudulent” (emphasis added) count as misbranding. But even then, in Seven Cases of Eckman’s Alternative v. United States (1916), the Supreme Court ruled that the language of claims needing to be both “false and fraudulent” meant that enforcement was only merited if the person selling the drug had intent to deceive. In other words, if the people selling the drug were mistaken but did themselves believe the efficacy claims they were making, then that was just a difference of opinion and FDA couldn’t crack down on them.
The FD&C Act in 1938 finally changed the definition of misbranding to include instances where “labeling is false or misleading in any particular,” and the courts upheld FDA’s ability to enforce on this basis. In Research Laboratories Inc. v. United States (1944), the 9th Circuit Court held that FDA was justified in seizing bottles of Nue-Ovo, a bogus treatment for rheumatoid arthritis. The court opined that scientific advances had made the McAnnulty decision obsolete in some cases:
Questions which previously were subjects only of opinion have now been answered with certainty by the application of scientifically known facts. In the consideration of the McAnnulty rule, courts should give recognition to this advancement.
This decision basically allowed FDA to crack down on efficacy claims under the “misbranding” provision of FD&C. The 1962 amendments established a different legal basis for efficacy regulation, though: they defined anything not yet approved or not yet recognized as safe and effective as being a “new drug” and required companies to apply to FDA for pre-market approval, presenting “evidence consisting of adequate and well-controlled investigations” (for discussion of what this phrase means, see “Clinical trial design” section below).
The 1962 amendments established a pre-market approval process only for “new drugs”. Drugs that were already marketed as of 1962 (and for which labeling, dose, etc. did not change) got grandfathered in, as did drugs that had already been grandfathered in 1938, and drugs “generally recognized as safe and effective” (GRASE). In practice, however, the Supreme Court ruled that “GRASE” status requires the same level of evidence as it would take for FDA to approve a new drug, and that the grandfather clauses only apply if the drug has the exact same labeling and composition that it had before 1962 (which is rare, as labeling usually evolves). Therefore in practice neither GRASE nor the grandfather clauses actually shelter any drugs from enforcement if FDA deems them ineffective. For decisions see Weinberger v. Hynson, Westcott & Dunning, Inc. (1973) and Weinberger v. Bentex Pharmaceuticals Inc. (1973). In lieu of subjecting every pre-1962 drug to the full NDA process (clearly outright infeasible), FDA implemented a (still gargantuan) process called the Drug Efficacy Study Implementation (DESI) program to evaluate the pre-1962 drugs. The DESI program is still ongoing today.
Access to unapproved drugs for desperately ill patients
Ever since 1962, debates have raged over whether FDA should be able to prevent patients with serious, life-threatening diseases from taking unapproved medicines.
One of the first major cases concerned amygdalin (marketed as Laetrile), a natural product purified from apricot kernels, then purported to possess anti-cancer properties, though clinical trials later failed to show efficacy [Moertel 1982]. Two cancer patients who wanted access to the drug sued the FDA. The West Oklahoma District Court initially ruled in their favor in Rutherford v. United States (1977), citing a constitutional right to privacy and arguing that allowing access would NOT mean “the return of the traveling snake oil salesman”, because “the right to use a harmless, unproven remedy is quite distinct from any alleged right to promote such”. The 10th Circuit Court upheld that decision but on a different basis, saying that the FD&C Act does not apply to terminally ill patients because “efficacy” is undefined when the alternative is certain death: “What can ‘effective’ mean if the person… is going to die of cancer regardless of what may be done.” The Supreme Court overruled both decisions, saying FDA absolutely had jurisdiction to keep those patients from taking amygdalin. In response, in 1977 Representative Symms led an attempt in Congress to overturn the efficacy provision of the FD&C Act, but was ultimately unsuccessful. Courts have since continued to uphold FDA’s power to keep seriously ill patients from accessing unapproved therapies, see for instance Abigail Alliance v. Von Eschenbach (2008). (Interestingly, the dissent in that case argued that if the constitutional right to Due Process covers the rights “to marry, to fornicate, to have children, to control the education and upbringing of children, to perform varied sexual acts in private, and to control one’s own body even if it results in one’s own death or the death of a fetus” then surely it must also cover the right to attempt to save one’s own life.)
Another major challenge came from the so-called buyers’ clubs during the HIV/AIDS epidemic, as dramatized in the film Dallas Buyers’ Club. According to this, FDA looked the other way for the first few years of the buyers’ clubs, but eventually cracked down.
The FDA Modernization Act of 1997 formalized a procedure for patients to request “compassionate use” of a drug that had Investigational New Drug (IND) status but was not yet approved. (In practice, FDA usually approves such requests, but drug companies are disincentivized against providing the drug, because FDA will consider adverse events encountered in compassionate use in its review of safety, yet asymmetrically, compassionate use instances do not contribute to efficacy data - citation needed.)
A recent development has been the introduction of Right To Try laws, which have passed in 28 states and have been proposed at the federal level. From a legal standpoint, however, these laws don’t do much, as all they say is that patients have a right to ask companies for investigational drugs (which in fact they always had), they don’t specify a right to receive the drugs.
Steps in new drug development
An Investigational New Drug (IND) application must be submitted and approved in order for a drug to begin first-in-human studies and enter clinical testing. Technically, the amended FD&C Act’s prohibition on interstate commerce on unapproved drugs would apply even to shipping a drug out to clinical trial sites in order to perform trials. The IND is the formal mechanism by which FDA exempts experimental new drugs from this prohibition.
As explained in FDA’s IND overview, an IND has three major parts:
- Animal studies. The IND needs to show that enough animal testing has been done to demonstrate that the drug appears safe to put into humans, and to choose a starting dose. At a minimum, the data package should include pharmacokinetics, pharmacodynamics, and toxicity, and FDA has fairly specific recommendations for how to structure the studies, in Guidance for Industry: Nonclinical Safety Studies (2010). Toxicity studies should be performed under Good Laboratory Practices (GLP) in two mammalian species, at least one of which is not a rodent. There should be enough study of the drug’s pharmacokinetics (what the body does to the drug), including ADME (absorption, distribution, metabolism, and excretion) to notice any worrisome metabolites and to identify potential organs of concern for toxicity. The drug’s mechanism of action should be identified and there should be enough study of pharmacodynamics (what the drug does to the body) to be sure that the animals in which you’re studying toxicity actually have relevant target engagement. Although target engagement data are expected, there is no requirement of preclinical efficacy studies per se (such as survival or other therapeutic benefit in a relevant animal model); the IND’s focus is on making sure the drug appears safe enough to warrant testing in humans.
- Manufacturing information. The FDA needs to be convinced that the applicant can produce the drug safely and reliably. Data should be presented on the “composition, manufacturer, stability, and controls”.
- Clinical protocols and investigators. The detailed protocol for Phase I (or Phase 0, see below) studies should be included — doses, timepoints, numbers, locations. It is recommended to include an IRB protocol and consent form; you don’t need to already have IRB approval but you need to at least commit to obtaining it. And you need to information about the study investigators. FDA’s goal is to make sure that investigators are qualified, research subjects will be appropriately informed and consented, and that no one will be exposed to undue risk. When Phase II and III studies are planned later, these are added as amendments to the IND.
INDs are approved by default if FDA does not respond within 30 days. Reasons why FDA might issue a hold on the IND include bad clinical trial design, unqualified investigators, or unreasonable risk to clinical trial participants.
After the IND, traditionally one would advance to a Phase I study. Since 2006, however, there is also a concept of a “Phase Zero” study, which FDA put forth in a Guidance entitled Exploratory IND Studies. The idea here is that animal models can only tell you so much, and so in some cases sponsors might find it useful to, very early in the development pipeline, do a very small study in humans to help decide, for instance, which of a few drug candidates is most promising. In such cases, FDA will accept an IND with less preclinical data than is usually expected, if the plan is to give only brief exposure to very low, subtherapeutic doses to humans. In such a study one would not expect to be able to evaluate efficacy, or safety, or even to identify the right dose, but one might be able to check a biomarker (say, drug distribution to a tissue of interest, or qPCR for a induction of a splicing product of interest) to see if a drug is going to the right place or engaging the right target.
The traditional clinical trial pipeline, especially for common diseases, involves three phases. The distinction between phases is less clear for rare diseases, where in many cases the design of trials is determined in case-by-case negotiations between the sponsor and FDA, more details below.
Phase I is traditionally thought of as a safety study, but data from Phase I are also instrumental in seeing whether the drug engages its target, whether it has good enough pharmacokinetics in humans, and to determine the dose and other parameters for a Phase II study of efficacy. Because Phase I only tests the drug’s safety in a small cohort over a short period of time, it won’t necessarily catch rare or long-term adverse events. Phase I is usually quite safe since the doses escalate gradually and subjects are closely watched for adverse events, but there are occasional and highly publicized disasters in which people die or become severely ill — see for instance Tegenero’s TGN1412 antibody [Suntharalingam 2006] or Biotrial’s BIA 10-2474.
Phase II is usually in a slightly larger cohort of people, still fairly closely monitored, and serves to generate preliminary evidence of efficacy.
Phase III is a larger and longer study that can evaluate the risk-benefit profile of the drug, hopefully confirming the results of the Phase II while at the same time having long enough exposure in a large enough number of people to get a better read on safety.
Starting from 1962, FDA generally required two “adequate and well-controlled investigations” (see next section for discussion of what this means) at the P < .05 threshold, but FDAMA in 1997 amended this to indicate that in some cases, one adequate and well-controlled trial would suffice [Hutt 2007]. In some cases, a Phase II trial is considered adequate enough, and a Phase III need never be conducted. Acceptance of only one trial, rather than two, usually requires P < .005 instead of P < .05. Particularly in rare diseases, there is a tremendous amount of flexibility (from FDA’s perspective) or uncertainty (from drug companies’ perspective) as to what evidence FDA will require in order to approve a drug.
Once the sponsor has done enough trials (often as pre-determined with FDA, see Special Protocol Assessments below) with sufficiently positive results, the sponsor submits a New Drug Application (NDA). An NDA typically includes the following sections in some form or another [Hutt 2007]:
- Manufacturing information
- Preclinical pharmacology + toxicity
- Human pharmacology
- Microbiology (what is this?)
- Clinical data
- Statistics
- Proposed labeling
NDAs tend to be quite long; I couldn’t find an example NDA online, but for eteplirsen, for example, even the NDA briefing document was 186 pages. FDA is not required to, but almost always does, convene an Advisory Committee to review NDAs, and it almost always follows the advice of the Advisory Committee. As review of an NDA is winding down and a drug appears to be nearing approval, the final few weeks tend to involve final negotiations over the exact labeling provided to doctors.
Clinical trial design
The FD&C Act says that drugs can only be approved based “substantial evidence” of efficacy from “adequate and well-controlled investigations”. FDA has defined “well-controlled” to mean that one of the following types of controls must be in place:
- Placebo control. Some patients get placebo and some get drug.
- Dose control. Different patients get at least two different doses of drug, again usually on a randomized, double-blind basis.
- No treatment control. Same as #1 but with no placebo. This might be used if there is good reason to not expect any placebo effect AND efficacy can be objectively measured.
- Active treatment control. Same as #1 but instead of placebo, some patients get standard of care (existing approved therapy).
- Historical control. Whereas #1 - #4 are all expected to be randomized and concurrent, there is sometimes an option to compare to historical data on untreated individuals. Usually historical controls aren’t as comparable to the treated patients as a randomized group would be, so FDA’s standards for accepting historical controls are high — they state that this option is reserved for “special circumstances”.
The concept of an “adaptive trial design” simply means that people can be shuffled around between the different arms of the study (e.g. between doses or between placebo and drug) after the study has started, in order to maximize statistical power to see an effect, without increasing the false positive rate [Bhatt & Mehta 2016].
Usually FDA wants to see a clinical endpoint (a measure of how a patient feels or functions) evaluated in trials, but in some cases a surrogate biomarker endpoint can be substituted — see more detailed discussion in my Accelerated Approval post though note that surrogate biomarker endpoints are not limited to Accelerated Approvals.
User fees and meetings between sponsors and FDA
The Prescription Drug User Fee Act (PDUFA) of 1992 established a system where drug companies pay FDA “user fees” for the review of their products to supplement appropriations from Congress. As of 2017, for instance, the price tag for a New Drug Application is $2.04 million. As of 2012, user fees made up 62% of FDA’s budget, and Congressional appropriations only 38%. While creating a user fee system, Congress and FDA have also enacted various mechanisms to enhance regulatory predictability and accountability by establishing deadlines and performance requirements for FDA review of drugs, and particularly, by establishing a system of regular, defined meetings between sponsors and FDA.
PDUFA is often credited with reducing wait times for FDA review. Here are FDA-published data from our textbook on how PDUFA relates to New Drug Application review times for new molecular entities (NMEs):
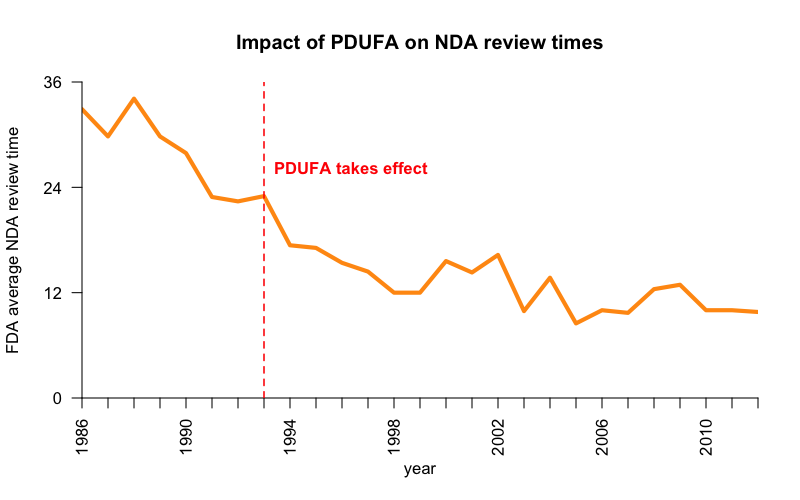
Code to produce this plot. Data from Food and Drug Law 4th Ed. p. 749.
There are a huge number of details to be hammered out in the design of clinical trials, and sponsors want to make sure that they are doing the right experiments and collecting the right data to ultimately win FDA’s approval. Therefore there is a formal structure for sponsors and FDA to meet regularly to discuss these details. The FDA Modernization Act (FDAMA) of 1997 further strengthened this process, adding requirements that minutes from these meetings be recorded and that FDA and sponsors can reach agreements in these meetings that will be written down and cannot be changed by FDA except in special circumstances. This is intended to provide regulatory predictability even if, for example, individual staff at FDA turn over, and is in contrast to the so-called “moving target syndrome” that sponsors had complained about before the 1990s.
During the drug development process, meetings are routinely scheduled pre- and post- preclinical studies, pre-IND, end of Phase I, end of Phase II, pre-Phase III, post-Phase III, and pre-NDA, plus Special Protocol Assessments (see next paragraph). 30 days before a meeting, the sponsor submits a briefing document, and they can include questions for FDA, each of which FDA will respond to 2 days before the meeting, often with a yes/no answer.
Most of these routine meetings are considered Type B meetings. If a meeting is needed to resolve a hold (see previous paragraph) or other roadblock, that’s a Type A meeting, and any other meeting related to drug development is Type C. FDA promises to schedule Type A meetings within 30 days of request, Type B within 60 days, and Type C within 75 days. (For a recent example, see Appendix 1 of Sarepta’s eteplirsen NDA briefing, p. 137-144, which lists all the key regulatory milestones including every meeting between Sarepta and FDA over eteplirsen). Some people have objected that these wait times are actually quite long for startup companies when you consider their burn rate.
Another mechanism intended to establish regulatory predictability is a Special Protocol Assessment (SPA). In this mechanism, a sponsor can submit a protocol for a Phase III trial that will form the primary basis for efficacy claims in an NDA, and FDA will respond within 45 days as to whether success in said trial design would merit approval. If FDA says yes, this is essentially binding. According to the 2002 Guidance on SPAs, FDA can break its promise only if it identifies “public health concerns unrecognized at the time of protocol assessment,” if the sponsor lied in its submission or failed to follow the protocol, or if “the director of the review division determines that a substantial scientific issue essential to determining the safety or efficacy of the drug has been identified after the testing has begun”. In practice, FDA has so far never once backed out on an SPA commitment.
Since 1962, after an NDA is submitted, FDA must decide whether to approve it within 180 days. In practice, however, FDA can request additional information, and then when that information is submitted, FDA can consider it a revision and thus reset the clock [Merrill 1996]. The 1962 amendments also provided that sponsors can challenge FDA’s rejection of a drug in court [Merrill 1996], but almost no one ever does (perhaps for the reasons explained in lecture 2), and not once has anyone ever done so successfully. For a rare example of an attempt, see Ubiotica Corp. v. FDA (1970).
Controversies
Controversy has long raged over whether FDA is too fast and loose (endangering consumers by approving drugs on too little evidence), or too cautious and slow (letting patients die by failing to approve drugs quickly enough). For a look at what this debate looked like 40 years ago, see [Kennedy 1978, Wardell 1978 ]. One fact that is not disputed is that “R&D efficiency”, defined as new molecular entities (NMEs) approved per research & development dollar spent, has declined by orders of magnitude over recent decades [Scannell 2012].
In spite of the improvements in NDA review time since PDUFA, overall time that a drug spends under an IND has increased over time:
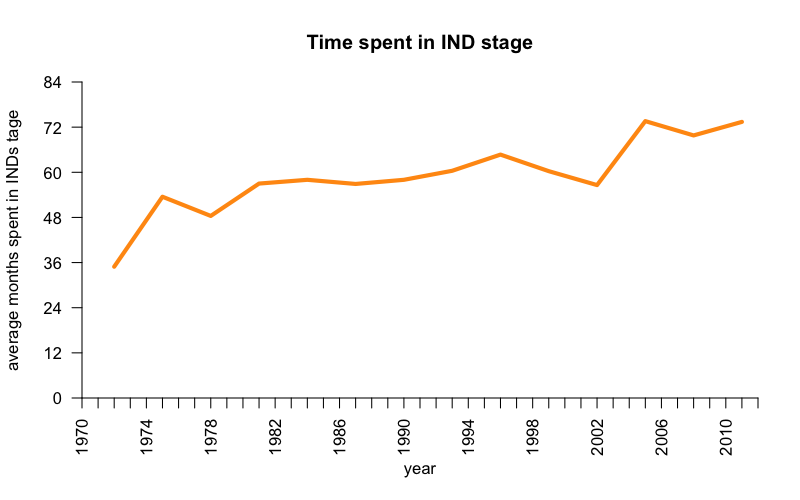
Code to produce this plot. Data from Food and Drug Law 4th Ed. p. 750.
Miscellanea
An FDA regulation, 21 C.F.R. 56.103(a), defines what an Institutional Review Board (IRB) is, and that any clinical trial performed under an IND has to be reviewed and approved by one, with all participants giving informed consent. There are very rare, specific exceptions to the informed consent rule, including emergency situations where the patient is unconscious and in terminal condition without any alternative treatment.
Historically, most clinical trials were conducted in middle-aged white men. Over the past 20 years, Congress and FDA have specifically enacted measures to require drugs to be tested in children, and there is now an exepctation of inclusion of women, elderly people, and people of other races in trials, with the statement that “Patients in clinical trials should, in general, reflect the population that will receive the drug when it is marketed” (58 FR 39410). There is one drug, BiDil®, approved and labeled only for African-Americans.
Trials increasingly have Data Monitoring Committees.
Trials can be conducted overseas and as long as study design and execution are consistent with its standards, FDA will view the data as being equivalent to a U.S. trial.
Since 1962, every imaginable type of investigator fraud has occurred in clinical trials, including just fabricating all of the data. Sponsors try to crack down on this by having Clinical Research Associates (CRAs) interview investigators periodically and review forms on patients, and FDA tries to crack down on this by disqualifying investigators who get caught, but no one has found a silver bullet to prevent all fraud.
FDAMA in 1997 required FDA to establish a public-facing database of clinical trials, a mandate that was implemented in the form of ClinicalTrials.gov. This was partly in response to pressure to make it easier for HIV/AIDS patients to be able to find trials they were eligible for. The site has been expanded and reformed over the years but continues to be heavily criticized for lack of enforcement (many trials, particularly NIH-funded ones, are not included), lack of detail (clinical trial protocols are not included), lack of results (only trials that later lead to drug approval are required to post results), and generally not being user-friendly enough.
There have been efforts to harmonize the drug approval process between FDA and its European Union counterpart, the European Medicines Agency (EMA).
The “intent-to-treat” population refers to all people who were initially randomized, even if they failed to comply with the treatment regimen, didn’t end up taking the drug or at least not in the dose they were supposed to, and thus could not possibly exhibit a benefit of the drug. FDA requires that the statistical analyses submitted to it for approval be based on “intent-to-treat,” so non-compliance can significantly water down the observed efficacy and thus the case for approval.
For antidotes or treatments for chemical toxins, biological agents, or radiation (anything from snake venom to bioterrorism), FDA can allow approval based solely on animal studies, without any human clinical trials. The rationale is that you could only study the thing by exposing people to the harmful agent in the first place, which would be unethical. (I’m not sure when/how this applies, as there certainly are human trials on people who’ve been exposed to something harmful: see intravenous milk thistle for Amanitas mushroom poisoning and the Ebola Ça Suffit! vaccine study [Henao-Restrepo 2016]).
Financial conflicts of interest (including working or consulting for pharma companies) are considered a bar to participation in an Advisory Committee, though FDA can grant waivers as long as it publicly discloses them. There have been various controversies and reform proposals regarding the conflict of interest policy.
NewCo, Part I
This is the first in a series of three episodes of a narrative about a fictional biotech startup company, NewCo, developing a new drug.
NewCo is a biotech startup developing a plant-derived natural product as a contrast agent for non-invasive “virtual” colonoscopies. It has a small amount of human data indicating the plant extract is well-tolerated.
The way colonoscopies work is that the patient has to do a “bowel prep” to clear out the digestive tract so that any cancerous polyps or other issues can be seen and doctors can remove them. For traditional, “optical” colonoscopies, the patient is anesthetized, and a scope is inserted into the rectum to take video. Complications are rare but serious, including risk of puncture (perforation) and even death. In a “virtual” colonoscopy, the patient still has to do the bowel prep, but then just takes a contrast agent and undergoes a CT scan; no scope is inserted. For background, see this Washington Post article. One virtual colonoscopy agent is already FDA-approved but it has less sensitivity for lesions <5 mm in diameter than optical colonoscopy, and many insurers do not cover it. There is also competition from a stool test for DNA and protein markers, but no agent has been shown comparable to optical colonoscopy. NewCo hopes its agent is better and can prove equally effective as optical colonoscopy.
To get approval for its agent, NewCo will need to compare performance to traditional, optical, colonoscopies. It will need to show at least comparable, if not better, image quality and detection sensitivity, including for relatively flat polyps which might be the hardest to detect. It may also help if the virtual colonoscopy (because it includes a full body CT scan) has the ability to generate life-saving incidental findings by detecting cancers in other tissues, although there is also a risk that these incidental findings will lead to unnecessary interventions and ultimately do harm. Because adverse events in optical colonoscopy are rare to begin with, NewCo probably won’t have statistical power in its small trial to prove that its virtual colonoscopy is safer than optical colonoscopy, but it will at least need data to suggest that it isn’t less safe: the CT scan does involve significant radiation exposure, which must be traded off against cancer prevention benefits of the procedure. And it will probably help if NewCo can also quantify the quality of life improvement for its patients as well. Many patients absolutely refuse optical colonoscopy, even when doctors strongly recommend it, so there is also a potential for benefit even if NewCo can just increase the percent of patients agreeing to undergo colonoscopy.
NewCo has raised a total of $20 million from two venture capital firms, and has a burn rate of $500K/month, meaning its “flameout date” (when it will run out of money at a constant burn rate) is 3 years and 4 months from now. Its burn rate will likely go up, however, as it advances preclinical and clinical studies. As a rule of thumb, biotech startups try to keep the flameout date always at least one year in the future.
The President and CEO runs the company; she may consult with the Board of Directors, the CFO, or others, but ultimately she makes all final decisions. The CFO is often someone who will later advance to being a CEO of this or another company, and has primary responsibility for making sure the venture capitalist investors are happy.
The active agent in the plant extract has not yet been identified, and there are questions about whether NewCo can patent the extract without identifying the active principle or developing a method for synthesizing the compound. Some within the company are advocating for $1 million per year to be devoted to compound identification and synthesis.
to be continued tomorrow…

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.