Antisense part III: chemistries
This post is third in a series of posts on the science of antisense oligonucleotides as therapeutics.
I began this series with an introduction to antisense oligonucleotides (ASOs) and their potential application to prion disease. My last post described the different mechanisms of action (MoA) that ASOs can have. The MoA of an ASO depends on its sequence — what position in the RNA it targets — but also on its chemistry. This post will cover in greater detail the different chemical modifications employed in ASOs, and what ends they can achieve.
First things first: for the non-chemists like me, it is important to review the structure of nucleic acid and how the positions are numbered:
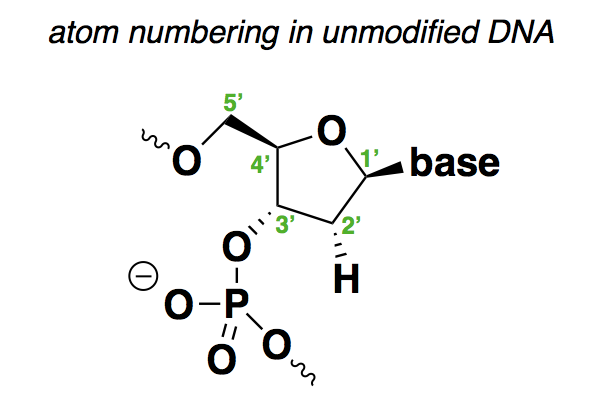
As shown above, the positions are as follows:
- 1’ is where the base (A, C, G, or T) is attached.
- 2’ is H in deoxyribonucleic acid, or OH in ribonucleic acid. The above structure contains many implicit hydrogens, including one at this very position, but I have drawn this particular hydrogen to show that it is what varies between DNA and RNA.
- 3’ is where the backbone connects to the next base.
- 4’ is where there is a branch off of the sugar ring to the 5’ position.
- 5’ is where the backbone connects to the previous base.
From this, you can see that the 1’, 3’, and 5’ positions in this molecule are pretty much spoken-for. Any chemical modifications are either going to have to be at the 2’ or 4’ positions, to the phosphodiester bonds in between the sugars, or are going to involve different sugar rings altogether.
Below is a non-exhaustive comparison of the chemistries that seem to crop up most often in drugs and in the literature, with the differences from natural nucleic acid highlighted in blue:
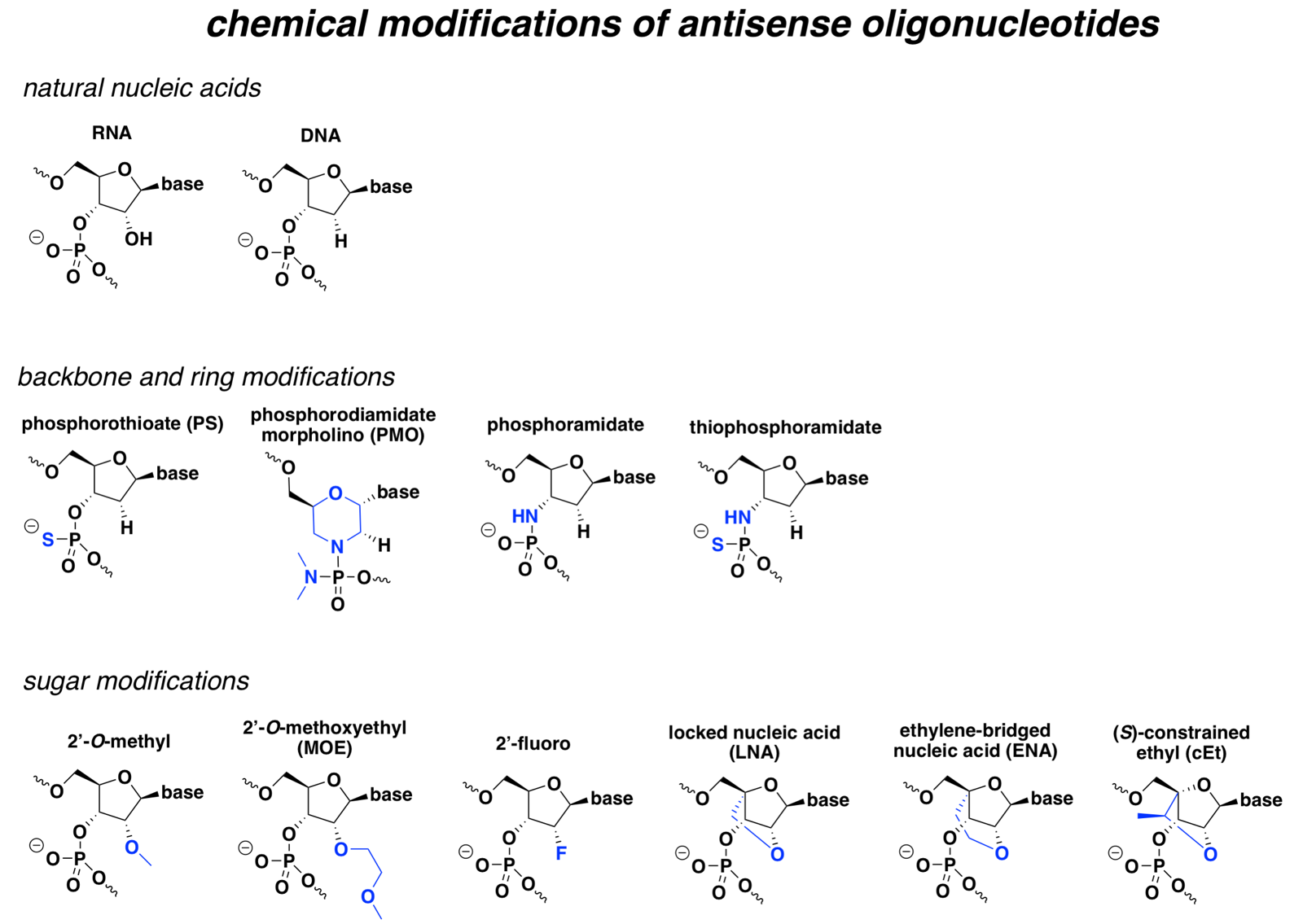
At broad strokes, most of these modifications improve either pharmacokinetics (improved nuclease resistance for a longer half-life), pharmacodynamics (improved affinity for the target RNA), or both, and some can also confer more specific special properties. But with the exception of PS, most also prevent cleavage by RNAse H, which is the desired MoA for many ASOs. Accordingly, many RNAse H ASOs are designed as chimeras, where the different bases are a mix of different chemistries, or as gapmers, where the “wings” have modifications that the central bases lack.
The sections below will explain the properties of each specific modification.
unmodified RNA and DNA
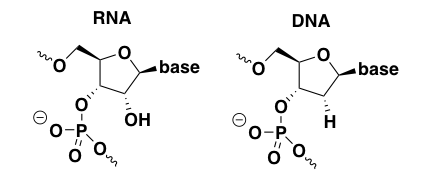
Unmodified, unformulated nucleic acids are seldom if ever used as drugs. Of the antisense and siRNA drug candidates in clinical trials as of last year, all were, if not chemically modified, at least formulated in some sort of lipid nanoparticle [Bennett 2017]. It is useful, however, to talk about the limitations of naked unmodified RNA and DNA in order to inform why all the chemical modifications below were invented.
It is difficult to find empirical data on unmodified DNA and RNA because no one thinks they have potential as drugs. But one review summarizes at least three main pharmacological problems with natural nucleic acids, any one of which might be sufficient to preclude their use as drugs [Bennett & Swayze 2010].
First, DNA and RNA are very quickly broken down by endogenous nucleases in the body. These include both exonucleases (which remove nucleotides from either end of a chain) and endonucleases (which cut in the middle of a chain).
Second, DNA and RNA have only minimal plasma protein binding. Most drugs bind to proteins in plasma, such as albumin, to some degree, and this is a huge determinant of the pharmacokinetics of the drug. In equilibrium, some drug is protein-bound and some is not. The protein-bound fraction of the drug circulates stably in the bloodstream, acting as a reservoir for drug to be released from the protein over time. It is the non-protein-bound fraction of the drug that’s active (it can be taken up into cells and engage its target), but it is also the non-protein-bound fraction that can be removed by the kidneys. Unmodified DNA and RNA apparently are excreted quickly in urine due to too little plasma protein binding — I couldn’t find any primary data for this, but a number of reviews make this claim — see for instance [Bennett & Swayze 2010, Geary 2015].
Third, while unmodified oligonucleotides might bind to an unstructured target RNA with acceptable affinity, the reality is that most RNA molecules have internal structures — they fold back on themselves in stems and loops — and a drug can only bind if it is so affine as to be able to outcompete that internal structure. I didn’t find any data on this phenomenon using natural DNA or RNA, but for one example that illustrates the problem using phosphorothioate oligos, see Table 1 of [Monia 1993]. In those experiments, a particular 17-base oligo could bind an unstructured target with Kd = 3 × 10-13 — that’s sub-picomolar affinity — but if the target was structured, the Kd rose above 1 × 10-5, or in other words, binding was not observed even at 10 μM. Therefore, one of the goals of chemical modification of oligos must be to improve their affinity for their target sequences to the point where they can compete with endogenous RNA structures.
phosphorothioate (PS)
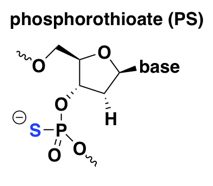
The PS modification is old and rather widely used, including in almost all of Ionis Pharmaceuticals’s ASOs in the clinic today. The first generation of ASOs to reach the clinic were simple, straight PS oligos — all linkages were PS and there were no other modifications. See for instance fomivirsen, which in 1998 became the first approved ASO drug, for cytomegalovirus retinitis (a complication of AIDS) [Perry & Balfour 1999]. More recent ASO drugs and drug candidates incorporate PS along with other modifications described below [Bennett 2017].
Compared to naked DNA, PS improves nuclease resistance — early in vitro experiments showed that DNAse digests of PS oligos were 2 to 45 times slower than their normal DNA (PO) counterparts [Stein 1988]. Yet fortuitously, as noted above, PS retains RNAse H-cleavability [Furdon 1989]. On the downside, apparently PS reduces the melting temperature of oligonucleotide duplexes [Stein 1988], or in other words, it decreases the affinity for the target RNA. This can be overcome by the use of 2’ modifications discussed further below, most of which improve affinity. PS also greatly increases the ASO’s protein binding [Brown 1994], which turns out to be important for two reasons. First, at least for peripherally delivered ASOs, as for any other systemic drug, plasma protein binding is important for allowing the drug to stay in circulation long enough to get taken up into tissue [Geary 2015]. Second, ASOs bind tens of different cellular proteins and many of these interactions are important for the uptake of ASOs into the cell and/or for their distribution to the right compartment within the cell [Liang 2015, Bailey 2017, Crooke 2017].
phosphorodiamidate morpholino (PMO)
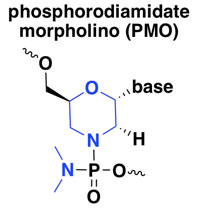
These ASOs are known simply as morpholinos, after the six-membered morpholine ring that substitutes for the usual five-membered ribose in nucleic acid, or as phosphorodiamidate morpholino oligonucleotides (PMO), with phosphorodiamidate referring to the replacement of two oxygens with nitrogen on the phosphate group. PMOs are fairly widely used, with several drugs or drug candidates in the clinic today, all of which are splice modulators [Bennett 2017]. I did find also one reference on a PMO against the oncogene MYC, a steric blocker of translation, that made it into a Phase I human trial [Iversen 2003, Devi 2005].
PMOs have improved nuclease resistance compared to normal DNA, however, they are also resistant to RNAse H [Hudziak 1996, Summerton 1999], effectively precluding their use for knockdown at the RNA level — instead, PMOs are used as splice modulators or translation inhibitors. Whereas the 2’ modifications listed below that confer RNAse H resistance can still be used in RNAse H oligos formulated as gapmers with cleavable bases in the middle, in a quick search, I did not find any papers or patents using a gapmer approach with PMOs. According to one review I read [Geary 2015], the fact that the PMO backbone is neutral (as opposed to natural nucleic acid and all other modifications discussed here, which are anionic) causes them to have lower plasma protein binding and thus quicker elimination from the body, which can make dosing more difficult.
In terms of mechanism of action, all the PMOs that have made it to human clinical trials [Bennett 2017] appear to be splice modulators, delivered intravenously for muscular dystrophy. As noted in the last post, PMOs also enjoy widespread academic research use in zebrafish as translation inhibitors, by a steric block mechanism, but this MoA has not been used much or maybe at all in rodents or humans.
The emerging clinical use in muscle disease does not mean that intravenous PMOs are readily taken up into muscle, at least not without huge doses. The FDA memo on eteplirsen, a PMO drug for DMD exon 51 skipping in Duchenne muscular dystrophy, noted (see p. 87 of the PDF) that the drug did not achieve appreciable exon skipping activity in monkeys until an IV dose of 320 mg/kg/week — that would mean a whopping 22 grams of drug per week in a 70 kg human. In its letter granting Accelerated Approval to eteplirsen, FDA stipulated a confirmatory study in which patients would be randomized to either the approved dose of 30 mg/kg/week or to a much higher dose of 30 mg/kg/day (210 mg/kg/week), closer to that pharmacodynamically effective dose in monkeys.
phosphoramidate and thiophosphoramidate
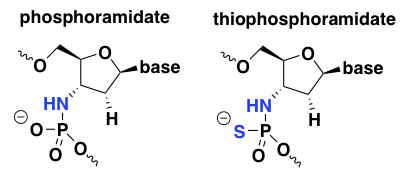
Much less work has been done on these two chemistries compared to PS and PMO. In a few searches I could not find any evidence that any ASOs with these chemistries are in human trials, or even that anyone is still working on them today, so I won’t spend too much time on them. Phosphoramidate oligos were reported to have good nuclease resistance and remarkably high duplex melting temperatures, suggesting excellent affinity for the target RNA in an antisense application [Gryaznov 1995, Gryaznov 1996]. However, they also had low protein binding and were susceptible to acid degradation, so thiophosphoramidate oligos were explored as an alternative [US6608036B1, Pongracz 1999].
2’-O-methyl
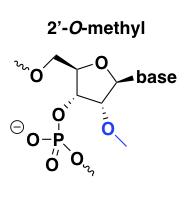
2’-O-methyl improves affinity for the target RNA, with perhaps a +0.6 to +1.2°C melting temperature shift per modified base [Monia 1993], and also improves nuclease resistance. It is usually used in combination with a PS backbone, and has good pharmacokinetics [Agrawal 1995, Zhang 1995].
This modification’s nuclease resistance comes at the expense of also being RNAse H-resistant. Thus, all bases can be 2’-O-methyl modified (“uniform” modification) if the desired mechanism is, say, splice modulation. Alternatively, the 2’-O-methyl modification can be used in a gapmer design to permit RNAse H cleavage. Indeed, what we now call a “gapmer” — an ASO with a particular chemical modification on the ends but not in the middle — was first demonstrated for 2’-O-methyl oligos [Inoue 1987, Shibahara 1987] and has been previously called a “chimeric splint” or an “RNAse H window”.
2’-O-methyl seems to have been one of the first 2’ modifications to be discovered and explored, so there are a lot of papers on it from the 1990s, but it is not too widely used today, perhaps because other 2’ modifications (see below) have proven superior in one way or another.
The only drug candidate I had heard of with the 2’-O-methyl modification was drisapersen, BioMarin’s candidate for Duchenne muscular dystrophy. Drisapersen had a PS backbone and every base was 2’-O-methyl modified; like eteplirsen (mentioned above) its goal was to cause DMD exon 51 skipping. Drisapersen was reported to have at least some promise based on early trials [Goemans 2011, Voit 2014] but ultimately failed to win FDA approval. BioMarin later ended development of drisapersen, along with follow-on ASOs in the pipeline for DMD exons 44, 45, and 53. I only found one example of a 2’-O-methyl gapmer with an RNAse H mechanism, an ASO against PRKAR1A that made it to a Phase I trial for cancer [Goel 2006].
2’-O-methoxyethyl (MOE)
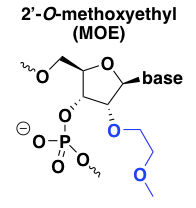
2’MOE chemistry has a number of advantages over the natural -H or -OH at this position in DNA or RNA. First, 2’MOE increases affinity for the target RNA, or the melting temperature of the duplex [Martin 1995, Altmann 1996, Teplova 1999]. Second, it confers almost total resistance to nucleases [Geary 2001]. In fact the nuclease resistance of 2’MOE is such that not all of the backbone phosphates need to be PS. Some of the 2’MOE nucleotides can be allowed to have normal PO linkages, without inviting nuclease degradation, and this mix of PO and PS linkages can be used to fine-tune the pharmacokinetics of the ASO, in some cases allowing more rapid distribution into tissue while keeping the terminal elimination rate slow [Geary 2001]. Finally, 2’MOE reduces the plasma protein binding of the ASO, which seems to shift its metabolism and excretion pathway away from the liver and instead towards the kidney for excretion in urine [Geary 2001].
Like 2’-O-methyl, 2’MOE is RNAse H-resistant, meaning it can be used in a uniform format for splice modulators, or in a gapmer design for RNAse H oligos. MOE is basically the lead chemistry employed in almost all of Ionis Pharmaceuticals’ ASOs currently in the clinic, both for CNS targets and for liver targets, and these examples include both uniform and gapmer designs.
Nusinersen, the approved splice-modulating ASO drug for spinal muscular atrophy, is uniform 2’MOE [Rigo 2014]. Mipomersen, the approved RNAse H oligo against APOB for familial hypercholesterolemia caused by homozygous LDLR deficiency, is 2’MOE gapmer. Specifically, it is a “5-10-5” oligo, meaning it has 5 MOE bases, 10 bases without a 2’ modification, and then another 5 MOE bases (all of the bases have a PS backbone) [Crooke & Geary 2013]. HTTRx, the ASO against HTT currently in trials for Huntington’s disease [Kordasiewicz 2012], and volanesorsen, the ASO against APOC3 currently in regulatory review for hypertriglyceridemia, are likewise 5-10-5 MOE gapmers.
2’-fluoro
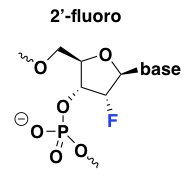
Of all the 2’ modifications, fluorine confers the strongest affinity for target RNA and has the strongest upward effect on the duplex melting temperature [Monia 1993, Freier & Altmann 1997], and thus, 2’-fluoro ASOs can be exceptionally potent. 2’-fluoro is also the most hydrophobic modification and the most intensely protein-binding [Liang 2016]. This modification also has the unique property, as explained in my MoA post, that it has the ability to promote exon exclusion by recruiting the ILF2/3 complex [Rigo 2012]. Unfortunately it turns out that 2’-fluoro oligos are also exceptionally toxic to the liver [Shen 2018], which is probably why there are no ASOs with this modification in clinical trials right now [Bennett 2017].
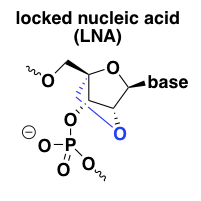
locked nucleic acid (LNA)
Understanding the LNA, ENA, and cEt modifications requires a bit of further background on the chemical nature of nucleic acid. The 5-membered sugar ring (ribose) is flexible and can shift dynamically back and forth between two possible conformations — one in which the 2’ atom is up and the 3’ is down (S or C2-endo), and the other in which the 3’ atom is up and the 2’ is down (N or C3-endo). This difference at the individual sugar ring level relates to the conformation of the nucleic acid chain as a whole, a topic introduced in molecular biology 04. C2-endo is the conformation of each ring in B-form (the normal conformation of DNA in a double helix), whereas C3-endo is the conformation of each ring in A-form (the conformation found in a DNA-RNA duplex). These conformations have very different geometries. Because ASOs target RNA and form a duplex with it, the desired conformation for target engagement is A-form — for instance, the crystal structure of RNAse H shows it binding an A-form DNA/RNA duplex [Nowotny 2007]. ASOs, even with PS modifications and 2’MOE and so on, remain flexible and can adopt either the C2-endo or C3-endo conformation. This flexibility limits their affinity for the target RNA, because each sugar ring is only in the right conformation half the time.
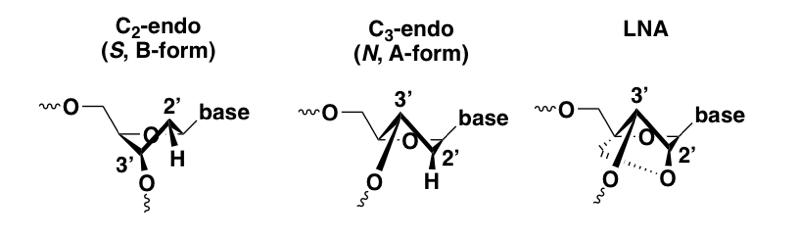
Conformational view of DNA sugar ring in the C2-endo and C3-endo conformation, compared with LNA.
Locked nucleic acid (LNA) was the first successful effort to add a reinforcement on the underside of the sugar ring to force it to stay permanently in the C3-endo conformation [Obika 1997, Wengel 1999]. The result is a profound affinity for the target RNA and thermal stability of the duplex, even stronger than the 2’-fluoro modification [Wahlestedt 2000]. For instance, a DNA-RNA duplex that melts at 59°C has no melting observed even at 90°C when the DNA is substituted for LNA [Wahlestedt 2000]. LNA is RNAse H-resistant but can be used in chimeric or gapmer ASOs. In keeping with the in vitro target affinity, some LNA ASOs were found to be much more potent in vivo than their 2’MOE analogs [Swayze 2007].
Unfortunately, LNA also turned out to have some liver toxicity issues [Swayze 2007]. The hepatotoxicity has been observed mostly in animal studies, but there is at least one clinical trial of an LNA that also reported liver function abnormalities [Bianchini 2013]. Evidently many but not all LNA oligos have this problem, and the molecular basis appears to lie in off-target degradation of some very long pre-mRNA transcripts [Burel 2016], an interesting story I’ll explore in more detail in a future post.
There are at least a couple of chimeric LNA drug candidates still in the clinic today [Bennett 2017]. Some are for systemic administration, such as miravirsen, a miR-122 antagonist delivered by subcutaneous injection [van der Ree 2016]. Others may avoid the liver toxicity issue by targeting a very specific tissue: for instance, ISTH0036, an LNA gapmer targeting TGFB2, is injected directly into the vitreous humor of the eye, and recently had acceptable safety profile reported from a Phase I trial [Pfeiffer 2017].
ethylene-bridged nucleic acid (ENA)
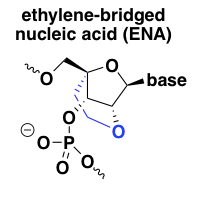
In light of the excellent target affinity and potency of LNA, chemists developed another, very similar, conformationally constrained nucleic acid analog called ethylene-bridged nucleic acid or ENA [Morita 2003]. It is just like LNA but with one additional carbon atom in the 2’ to 4’ linker. From my limited Google Scholar searching, ENA seems less extensively characterized in vivo than LNA at this point, but in vitro, its properties seem to be very similar to LNA [Koizumi 2004]. I found only one ENA ASO currently in clinical trials, Daiichi Sankyo’s DS-5141b, a splice-modulating oligo against DMD exon 45 for Duchenne muscular dystrophy (NCT02667483). There are no publications on it yet, but a conference poster described its pharamcological properties in mice as “stunning”, with significant exon skipping in heart muscle apparently observed even at doses as low as 3 mg/kg/week [Takaishi 2017]. If true, that’s 100 times lower than the dose required for good exon skipping activity in muscle in primate studies of eteplirsen (see discussion under PMOs above).
(S)-constrained ethyl (cEt)
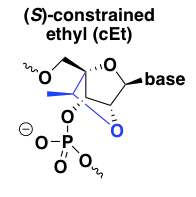
cEt is pronounced as two syllables, “SEE-et”. This is the most recent iteration on the concept of locking the 2’ and 4’ positions together to constrain the ribose ring into a C3-endo conformation [Seth 2008, Seth 2010, Pallan 2012]. Its target affinity is reported to be similar to that of LNA, while its degree of nuclease resistance is truly heroic, far outpacing both LNA and 2’MOE, which are themselves much better than normal DNA [Seth 2010]. Like LNA, it is RNAse H-resistant and so needs to be used in a gapmer design. The first paper on cEt made the claim that cEt oligos have a better tolerability profile than LNA, but no data were presented [Seth 2008]; detailed tox studies on one cEt gapmer clinical candidate reported that the tox profile was similar to 2’MOE ASOs [Burel 2013]. The more recent report attributing the mechanism of LNA hepatotoxicity to off-target RNA cleavage [Burel 2016] found that cEt oligos had similar properties as LNA, but that safe oligos could be identified through intensive preclinical screening.
A number of cEt gapmers or mixed MOE/cEt gapmers have had preclinical development reported — see for instance the one targeting DMPK for myotonic dystrophy [Pandey 2015]. And a few have made it to human clinical trials — see for instance a trial of a cEt gapmer targeting STAT3 in cancer patients, reported to have good potency and tolerability [Hong 2015]. As far as I can tell, none have made it into the human CNS yet. A recent paper described two new ASO candidates for SOD1 ALS [McCampbell 2018] as improvements over the original SOD1 oligo [Smith 2006, Miller 2013], but it looks like of the two new candidates, it was the MOE gapmer (ASO 1), not the mixed MOE/cEt gapmer (ASO 2), that has advanced to the clinic in NCT02623699 [McCampbell 2018].
comparison of chemical modifications
The below table summarizes the effects of each modification, compared to normal DNA. Definitions and considerations:
- Tm: melting temperature, where higher is better and means stronger affinity (lower Kd).
- nuclease resistance: half-life or time required for nucleolytic degradation, higher is better.
- protein binding: higher means more ASO is expected to be protein-bound. higher is better, at least to a point.
- RNAse H: does the modification support RNAse H cleavage at modified bases? (If not, gapmer designs are still viable.)
| modification | Tm | nuclease resistance | protein binding | RNAse H | comments |
|---|---|---|---|---|---|
| phosphorothioate (PS) | ↓ | ↑ | ↑ | ✔ | |
| phosphorodiamidate morpholino (PMO) | ↑ | ↓ | ✘ | ||
| phosphoramidate / thiophosphoramidate | ↑ | ↑ | ↓ | ✘ | susceptible to acid degradation |
| 2’-O-methyl | ↑ | ↑ | ✘ | ||
| 2’-O-methyoxyethyl (MOE) | ↑ | ↑ | ↓ | ✘ | allows mixed PO/PS backbone |
| 2’-fluoro | ↑↑ | ↑ | ↑ | ✘ | can promote exon exclusion |
| locked nucleic acid (LNA) | ↑↑↑ | ↑↑ | ↓ | ✘ | |
| ethylene-bridged nucleic acid (ENA) | ↑↑↑ | ↑↑ | ↓ | ✘ | |
| (S)-constrained ethyl (cEt) | ↑↑↑ | ↑↑ | ✘ |
stereochemistry
In the chemical structures at top, I drew all the bonds in the phosphate backbone with plain lines. But this ignores a chemical property that emerges when backbone substitutions are made: chirality. While a normal DNA backbone is non-chiral, any base with phosphorothioate (PS) or phosphorodiamidate (PMO) substitutions has a phosphorous atom bonded to four different groups, and that means that atom is a chiral center. The two different enantiomers of PS DNA, for example, are shown below:
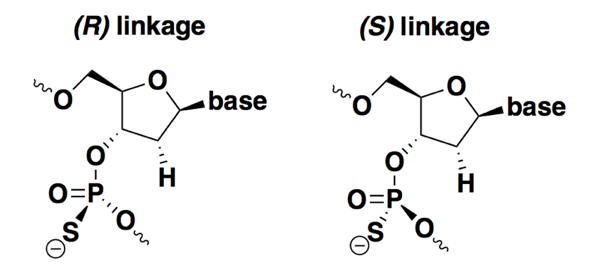
Most ASOs in development or in use as drugs today are stereorandom — they are synthesized with no control over stereochemistry, and so the drug exists as a collection of 2n-1 diastereomers, where n is the number of bases. Methods were developed long ago for synthesizing stereopure PS oligonucleotides, but only for all-Rp or all-Sp configurations [LaPlanche 1986]. It wasn’t easy to study any specific combinations or sequences of R and S linkages because it wasn’t easy to chemically synthesize them. In the past decade, and particularly the past few years, there has been an explosion of chemical synthesis methods for controlling chirality at each base of an ASO [Oka 2008, Nukaga 2016, US20150211006A1, EP3331891A2, Knouse & deGruyter 2018]. Therefore, in addition to a body of literature that has looked at differences between all-Rp and all-Sp oligos [Stec 1997, Li 2017], there have now been a couple of studies asking whether any specific stereoisomers have improved properties compared to stereorandom ones [Wan 2014, Iwamoto 2017].
Overall from the literature, it is clear that Rp and Sp linkages differ in a few key properties, but there is not yet any agreement on whether it is possible to design stereoisomers that are clearly superior to stereorandom ASOs.
In terms of different properties, compared to stereorandom oligos, each Rp linkage slightly increases melting temperature (meaning they improve affinity) while Sp linkages slightly decrease it [Koziolkiewicz 1995, Wan 2014, Li 2017, Iwamoto 2017]. On the flip side, Rp linkages appear to be degraded more quickly either in vivo or in serum or liver homogenate, whereas Sp are more stable [Wan 2014, Iwamoto 2017]. As for promoting RNAse H activity, it seems that both pure Rp and pure Sp oligos may be slightly poorer human RNAse H1 substrates than stereorandom ASOs [Wan 2014, Iwamoto 2017]. So far, it’s clear that there are tradeoffs: neither Rp nor Sp has all the properties you want in a drug.
If a good ASO will need a mix of some Rp and some Sp linkages, is there a particular sequence or combination that is optimal? Here, the findings have differed and there is no consensus yet. One study synthesized the same ASO nucleotide sequence with 29 different randomly selected sequences of Rp and Sp linkages, and did not find any that had better potency than the stereorandom equivalent in cell culture [Wan 2014]. Another study reported that a specific repeated stereochemical motif — SSR — improved RNAse H activity in a cell-free assay, but they found no difference in potency in cell culture [Iwamoto 2017]. In vivo experiments for a couple of ASO sequences, however, did show some evidence for longer-lasting effects with an SSR backbone compared to a stereorandom one [Iwamoto 2017]. This is a new area of research and it remains to be shown whether stereopure synthesis has any benefits that will generalize across ASO sequences.
quick final summary
Unmodified DNA and RNA have poor pharmacokinetics: they have insufficient affinity for their targets, are quickly broken down by nucleases, and have low plasma protein binding leading to swift excretion. A variety of chemical modifications have been invented to improve these properties. Phosphorothioate (PS) or phosphorodiamidate morpholino (PMO) backbones are often used. Modifications at the 2’ position of the sugar can improve affinity, protein binding, and nuclease resistance. Modifications spanning the 2’ and 4’ positions to conformationally constrain the sugar can give dramatic improvements in affinity. All modifications except PS prohibit RNAse H cleavage, but this can be rescued by a “gapmer” design where the modification is found only in the “wings” of the oligo, and the central bases are just PS.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.