Antisense part II: mechanisms of action
This post is second in a series of posts on the science of antisense oligonucleotides as therapeutics.
In my last post in this series I talked about the basics of what antisense oligonucleotides (ASOs) are, and why they seem promising as a therapeutic strategy for prion disease. This post will discuss their mechanisms of action (MoA) in more detail.
ASOs are designed to bind a target RNA through Watson-Crick base pairing: C-G, A-T. They can then modulate the stability, processing, or activity of that RNA through a number of different mechanisms [Bennett 2017]. I’ll break these into four clearly defined categories of effects the ASO can have, plus a grab-bag of “Other”:
- RNA knockdown. ASO recruits RNAse H to cleave and degrade the RNA, lowering RNA levels and thereby lowering protein levels.
- Splice modulation. ASO causes an exon to be preferentially included or excluded.
- Inhibiting translation. ASO sterically blocks translation machinery, lowering protein but not RNA levels.
- Increasing translation. ASO blocks upstream open reading frames (uORFs) or other inhibitory elements in the 5’UTR, increasing translation efficiency.
- Other. There are also a handful of other, less clearly characterized mechanisms of action which I will cover briefly.
The vast majority of the ASOs in clinical trials in humans today, of which there are tens, have one of the first two effects: either RNA knockdown or splice modulation [Bennett 2017]. The steric block mechanism is, as far as I can tell, mostly used as a tool in zebrafish [Bill 2009], and the discovery that ASOs can increase translation is still pretty recent [Liang 2016, Liang 2017b]. Here I will cover these four mechanisms with a bit of detail and then very briefly touch on some things in the “Other” category.
What determines the mechanism of action of an ASO? It appears to be a function mainly of two variables: sequence and chemistry. The sequence determines where in the RNA the ASO will bind. The chemistry determines whether it is RNAse H-cleavable, as will be reviewed in more detail in the next post. Here, I’ll go through each of these mechanisms one by one, and then at the end I’ll have a table comparing them.
1. RNA knockdown
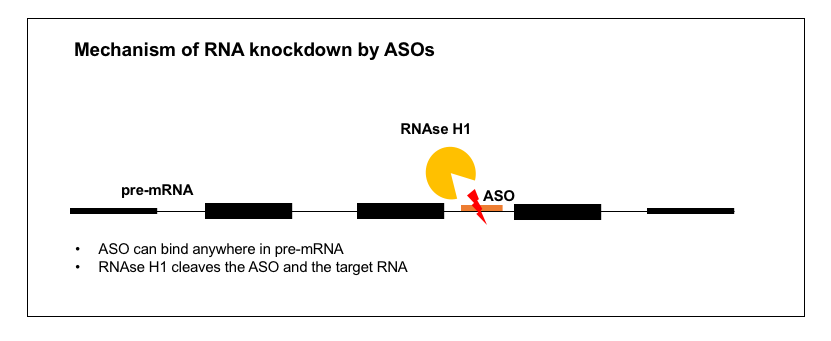
There are a great many diseases where what you want is to lower the abundance of one specific RNA and therefore its specific protein product. This is the case for prion disease and for many other gain-of-function diseases — for just a few examples in the CNS, see Ionis’s ASOs for HTT (Huntington’s disease) [Kordasiewicz 2012], SOD1 (ALS) [Smith 2006, Miller 2013, Winer 2013, McCampbell 2018], C9orf72 (ALS) [Lagier-Tourenne 2013], MAPT (tauopathies) [DeVos 2017], and ATXN2 (spinocerebellar ataxia type 2) [Scoles 2017]. There are also examples where the disease is loss-of-function but you can alleviate it by knocking down a different gene in the pathway — for instance, the approved ASO drug mipomersen targets APOB in the liver to reduce LDL in people who are lacking LDLR [Raal 2010], and the drug volanesorsen, currently awaiting regulatory decisions from FDA and EMA, targets APOC3 to lower triglycerides in people lacking LPL [Gaudet 2014].
In all of these cases, we know that the mechanism of action is lowering of the target RNA, rather than inhibition of translation, because the ASOs were discovered through qPCR-based cellular screens. For a bit about how these screens work, see the “Strategy” section and Figure 1 of the paper on Ionis’s former collaboration with the Prusiner lab on PRNP ASOs [Nazor Friberg 2012], or the associated patent [US20110269818A1]. As far as I could tell from the reading I did, ASOs to lower RNA levels do not require any specific sequence motif or any specific functional location within the targeted RNA molecule, but nonetheless, finding a reasonably potent ASO is non-trivial — it cannot be predicted informatically and does require screening.
How exactly do these ASOs lower their target RNA molecules? Decades ago, it was already suspected [Uhlmann & Peyman 1990] that the mechanism of RNA-lowering ASOs might rely on an enzyme called RNAse H, which had been found in cell-free systems to cleave RNA-DNA duplexes but not RNA-RNA duplexes [Minshull & Hunt 1986]. Conclusive evidence for the role of RNAse H (specifically the human enzyme RNAse H1, encoded by the gene RNASEH1) has come more recently. It was found that overexpression of RNAse H1 (in cell lines or in mouse liver) increased the potency of ASOs, meaning, the percent knockdown was increased for any given dose [Wu 2004]. Because RNAse H1 is essential [Cerritelli 2003], the converse experiment, of constitutively knocking out the gene, couldn’t be done. But conditional knockout of RNAse H1 in the mouse liver using a Cre system was found to abolish the activity of ASOs in that tissue [Lima 2016]. Immunocytofluorescence studies have shown ASOs to be localized to both the cytoplasm and the nucleus [Crooke 2017], and it turns out that RNAse H1 is active in cleaving ASO-RNA duplexes in both of these compartments as well [Liang 2017a]. Thus, RNAse H-mediated ASOs can target introns as well as exonic or untranslated regions of an RNA molecule, and some act exclusively through cleavage of pre-mRNA in the nucleus — see for instance the kinetics of RNA knockdown by a SOD1 intron-targeting oligo in Figure 6 of [Liang 2017a].
An incredible amount of literature exists on RNAse H1, and I only managed to scrape the surface. There is a crystal structure (PDB 2QK9) of RNAse H1 bound to a DNA-RNA duplex [Nowotny 2007], and a lot is known about its preferred cut sites [Wu 1999], functional domains [Wu 2001], and tolerance/preference for different chemical substitutions on the ASO backbone [Lima 2007]. It was counterintuitive to me that RNAse H should be active in the nucleus, because isn’t the nucleus full of natural DNA-RNA duplexes that arise during transcription, and how does the cell make sure these don’t get cleaved at random? I didn’t find a clear answer to this in any of the papers I read, but it is generally held that RNAse H1 actually has a native function involved in DNA replication. I didn’t find any references with a clear delineation of its native function in the nucleus, but the embryonic lethal phenotype in mice appears to arise from a defect in mitochondrial DNA replication [Cerritelli 2003], and recessive hypomorphic missense mutations in humans likewise cause a disease of mitochondrial DNA replication deficiency [Reyes 2015]. Regardless of what RNAse H1 normally does, the evidence is clear that RNA-lowering ASOs act through RNAse H1 and are active in the nucleus.
2. Splice modulation
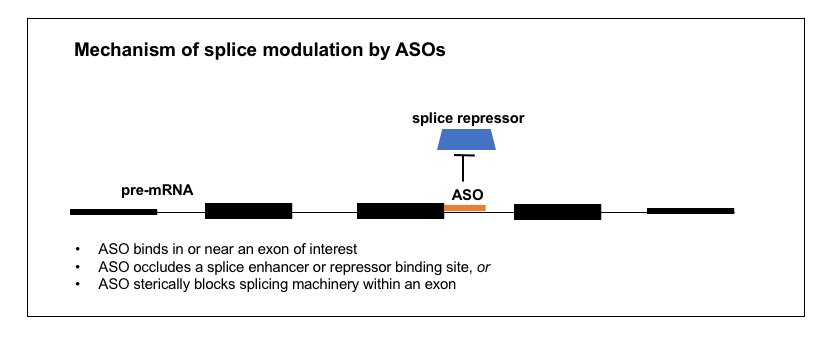
A number of ASOs in the clinic today are splice modulators. Nusinersen (formerly known as ISIS 396443), the approved ASO drug for spinal muscular atrophy, targets the SMN2 pre-mRNA in order to induce exon 7 inclusion and thus increase production of a full-length SMN2 protein that can substitute for the SMN1 protein that is missing in SMA patients [Finkel 2017]. Eteplirsen for Duchenne muscular dystrophy [Mendell 2013] targets the DMD transcript to promote exclusion of exon 51 in the subset of Duchenne patients where this either skips over their mutation or restores the proper reading frame [Aartsma-Rus 2009]. Several other splice-modulating ASOs are in trials right now, mostly for various exons of DMD [Bennett 2017]. Thus, ASOs can be designed to promote either the inclusion or exclusion of an exon of interest.
How does an ASO bring about a change in splicing? I took a close look at the development of nusinersen to try to understand the process. The goal was to cause SMN2 exon 7 to be included in the mature mRNA. A study had identified a “silencer” (intronic splicing silencer N1 or ISS-N1) in intron 7, just past the end of exon 7 [Singh 2006]. The “silencer” was just 15 nucleotides long and it started 9 nucleotides after the end of exon 7. Deleting or mutating it dramatically increased exon 7 inclusion, and binding it with an ASO mimicked this effect [Singh 2006]. Ionis screened ASOs targeting different positions within exon 7 [Hua 2007] and then in the surrounding intronic regions [Hua 2008] and they were able to identify very potent ASOs targeting the silencer [Hua 2008]. By looking at what proteins were bound to the RNA and by doing overexpression experiments in cells, they were able to determine that the silencer was a binding site for proteins hnRNP A1 and A2, which are splice repressors (they prevent exon inclusion), and the ASOs targeting this site prevent them from binding [Hua 2008]. The ASOs proved effective in mouse models of SMA [Hua 2008, Hua 2010] and soon went on to detailed pharmacological studies [Rigo 2014] and then the clinic [Chiriboga 2016, Finkel 2016, Finkel 2017, Mercuri 2018].
Nusinersen, then, acts by binding an intronic silencer in the pre-mRNA, and occluding a splice repressor protein from binding there. The mechanism of ASOs like eteplirsen, that promote exon exclusion, is less clear. One study found ASOs to 15 different DMD exons could all result in exon skipping, regardless of any obvious sequence motifs or splice site characteristics [Aartsma-Rus 2002]. The authors speculated that simply binding anywhere in the exon may interfere with the splice machinery [Aartsma-Rus 2003]. There are probably a variety of other mechanisms by which an ASO could modulate splicing too. For example, one chemistry of ASO (2’-fluoro) was found to recruit the ILF2/3 protein complex, thus promoting exon exclusion [Rigo 2012].
Regardless of exact mechanism, all of the splice-modulating ASOs I was able to read about acted locally rather than distally, meaning, they were targeted to a sequence in or near the exon of interest. Thus, finding a splice-modulating ASO involves a more constrained search space than finding an RNAse H ASO. Just as above, the method for finding them involves a qPCR screen in cultured cells, though you need primers spanning an exon junction with the exon of interest (and in the case of nusinersen they also needed a restriction enzyme digestion step to distinguish SMN2 from SMN1) [Hua 2007, Hua 2008]. Obviously, splice modulating ASOs can only work if they are active in the nucleus, and they can bind introns. A number of ASO backbone chemistries are resistant to RNAse H cleavage [Bennett 2017] and these can be used to make sure your would-be splice-modulating ASO doesn’t inadvertently result in knockdown. ASO chemistries will be reviewed in greater detail in the next post in this series.
3. Inhibiting translation
![]()
By binding a target RNA, an ASO can prevent the ribosome from translating the RNA, and thus reduce protein level without reducing RNA level. This is known as a “steric block” of translation. As far as I know, there are no approved drugs nor drug candidates currently in clinical trials that are translation-blocking ASOs [Bennett 2017]. Indeed, this mechanism of action seems little used in mammals: so far I have found only a few examples of translation-blocking ASOs that have been tested in rodents or pigs [Arora 2002, Kipshidze 2002,McCaffrey 2003], and just one Phase I trial in humans [Iversen 2003, Devi 2005]. Instead, this mechanism seems to be used chiefly in zebrafish, where it is a common research tool for doing gene knockdowns [Nasevicius & Ekker 2000, Bill 2009]. The preferred chemistry here is a phosphorodiamidate backbone (also known as morpholino or PMO).
Intrinsic to this mechanism of action is that the ASO’s activity is in the cytoplasm, where translation occurs, as opposed to in the nucleus, and the ASO must target a site in the mature mRNA. Indeed, apparently most steric blocking ASOs target a sequence either in the 5’UTR just upstream of the start codon [Faria 2001] or within ~25 bases past the start codon [Summerton 1999]. Unlike with qPCR for RNAse H or splice modulating oligos, there is no generalizable method for empirical screening of steric blocking oligos — one would need a different protein-based quantification method for each target of interest. From what I could gather online, it seems that most researchers are not doing extensive screening of steric block ASOs — one primer suggests that “if available, the level of knockdown should be assessed using an antibody to the protein of interest” [Bill 2009], implying that reseachers routinely buy steric block ASOs that have been synthesized based on informatic predictions alone, and that empirical demonstration of knockdown is left as an exercise.
There has been some recent controversy in the zebrafish community about how much people should or shouldn’t rely on steric block ASOs as research tools [Blum 2015, Stainier 2015], because one major study found that they failed to phenocopy genetic mutants across a range of different genes [Kok 2015]. Some current guidelines suggest that these compounds should be used less in isolation and more in conjunction with genetic tools such as CRISPR [Stainier 2017].
4. Increasing translation
![]()
There are a great many human diseases where the therapeutic goal would be to increase protein abundance, so if ASOs targeting uORFs could achieve this, they might have broad applicability. Work on this mechanism is still nascent but two recent papers suggest a couple of ways this could be achieved.
The first mechanism, involving upstream open reading frames (uORFs), is complicated and requires a bit of background. The translation rate of certain human genes can be regulated by upstream open reading frames (uORFs), as mentioned previously on this blog here and here. The mechanism is as follows. In order to properly translate an mRNA, the ribosome must associate with the canonical start codon (AUG) for the correct open reading frame. If there are other AUGs upstream of, and out-of-frame relative to, that canonical AUG, then ribosomes may initiate translation at those AUGs instead of the canonical one. This translation is not only non-productive (it doesn’t translate the in-frame amino acid sequence) but also competitive (the ribosomes that are already midstream translating out-of-frame can block the association of other ribosomes to the in-frame canonical AUG). The canonical example of a gene regulated in this manner is ATF4 [Harding 2000, Vattem & Wek 2004], whose translation rate paradoxically increases at times when translation of other transcripts is suppressed in response to cell stress, thanks to reduced initiation at its uORFs. But more recent evidence has shown that a large fraction, perhaps about half, of all human genes have uORFs that affect translation efficiency [Calvo 2009, Barbosa 2013]. In the recent paper on ASO targeting of uORFs [Liang 2016], they designed ASOs against four different genes’ uORFs, and were able to achieve +30% to +150% increase in protein abundance. Funnily enough, one of the targets was RNASEH1, perhaps chosen because the authors had abundant experience quantifying RNAse H1 from their studies of its role in ASO activity (see above).
The other mechanism, reported last year, simply goes after other translation-inhibitory elements in the 5’UTR, chiefly large stem-loop structures [Liang 2017b]. Some RNA molecules have particularly long 5’UTRs that fold back on themselves, forming stems and loops that somehow inhibit translation, perhaps just because there is less single-stranded RNA for translation initiation factors to bind. By targeting these inhibitory structures, the authors were able to achieve up to +170% increases in protein level, including for therapeutically relevant targets such as LDLR.
As far as I could find, all this work is still in the early mechanistic stages — I did not find any drug candidates in preclinical or clinical development with these mechanisms.
5. Other mechanisms
There are probably many other mechanisms out there, either waiting to be discovered, or already discovered and waiting for me to read about them. But I’ll just briefly mention a few things in the “other” category.
One really old review I read [Uhlmann & Peyman 1990] claimed that inhibition of transcription was another mechanism of ASOs. But it didn’t give much detail, and they only cited two examples. One was in bacteria and cited as conference talk with no paper associated; the other was of the human oncogene MYC but was purely based on an in vitro system [Cooney 1988]. None of the more recent reviews that I read mentioned a transcription-inhibiting mechanism, so I am guessing this idea either didn’t validate in vivo or at least hasn’t been developed further.
Another set of mechanisms involves micro-RNA. micro-RNAs or miRs are naturally occurring 22-nucleotide RNA molecules that bind the 3’UTRs of mRNAs in an antisense fashion. They were first discovered in C. elegans [Lee 1993, Wightman 1993], as reviewed in BBS 230, and are now known to be present in humans as well. miRs generally act to decrease the abundance of the protein encoded by the mRNA they target. miRs themselves act by a variety of mechanisms, including directing RNA cleavage (by Argonaute, the same protein responsible for siRNA activity), destabilizing the mRNA or reducing its translation rate [Bartel 2009]. It seems natural, then, that synthetic ASOs might aim to either mimic or block the activity of natural miRs, and indeed, ASOs have been developed to do both of these things. To my knowledge there are no approved drugs, but one drug candidate in clinical trials, miravirsen, is directed against human miR-122 and is being tested against hepatitis C [van der Ree 2016]. For some in vivo data, see the ASO against miR-33 for atherosclerosis [Rotllan 2013]. Yet another ASO, MRDX34, is not an antagonist but rather a mimetic of miR-34a and is being tested in cancer [Beg 2017]. I didn’t find a ton of papers on the exact mechanism of these ASOs, but it may not always be as simple as the ASO acting as, or binding, the miR. One study found that miravirsen not only binds miR-122, it also binds its precursor (pri-miR-122) and prevents its processing by Drosha (the enzyme that cuts a miR out of a longer RNA sequence), thus reducing the amount of miR-122 [Gebert 2014].
Many reviews of ASOs include short interfering RNAs (siRNA), since after all, siRNAs are also strings of nucleotides designed to be reverse complement to a gene of interest, and thus qualify as antisense oligonucleotides. I’m mostly excluding siRNA from this and other posts in this series, but for completeness I will briefly mention them here. Like many ASOs, siRNAs result in RNA knockdown, but they do so via a different mechanism independent of RNAse H, instead relying on the complexes Dicer, RISC, and Argonaute [Corey 2007, Davidson & McCray 2011]. Many siRNA drugs are in clinical trials and one, the peripherally delivered patisiran targeting TTR to treat hereditary transthyretin amyloidosis, appears to be close to approval [Morrison 2018].
Because I’m focusing in this blog post on antisense oligonucleotides, meaning, those designed to be reverse-complementary to a targeted gene, I won’t spend much time on aptameric mechanisms of action, meaning, things that oligonucleotides can do by directly binding proteins rather than by binding RNA in a sequence-dependent manner. But there is at least one approved drug that is an oligonucleotide with an aptameric mechanism — pegatinib, approved for macular degeneration, is a PEG-conjugated 28-mer oligonucleotide that binds and blocks the activity of VEGF. And it’s worth also mentioning that multiple studies have found that phosphorothioate oligonucleotides (PS oligos), a backbone chemistry often used in ASOs, bind PrP in a sequence-independent manner and can reduce prion load in cell culture [Kocisko 2006, Karpuj 2007, Nazor Friberg 2012]. The Prusiner lab argued that this “complicates any interpretation” of their ASO studies in prion-infected mice [Nazor Friberg 2012], because they were uncertain whether RNAse H-mediated PrP knockdown, or an aptameric mechanism, mediated the in vivo benefit they observed. We will address this question in future publications on ASOs for prion disease.
Finally, there are lots of other cool things ASOs can do, but as far as I can tell, most ultimately boil down to one of the above-listed mechanisms. For example, an ASO developed for Angelman syndrome upregulates UBE3A RNA, but it does so by targeting the long non-coding RNA (lncRNA) that suppresses UBE3A, using a conventional RNAse H mechanism [Meng 2015].
Comparison
Here is a table comparing some key attributes of the above mechanisms of action.
| effect | target | screening strategy | MoA |
|---|---|---|---|
| RNA knockdown | pre-mRNA; can be introns, exons, or UTRs | qPCR screen for RNA abundance | RNAse H1 cleavage |
| splice modulation | pre-mRNA; can be specific splice enhancers or silencers, or just exons | qPCR screen with primers spanning exon junction | splice enhancers/suppressors such as hnRNP A1/A2, or possibly steric block of splice machinery |
| inhibiting translation | 5’UTR or first exon, near start codon | none generalizable; requires protein quantification assay | steric block of translation machinery |
| increasing translation | 5’UTR stem-loops or uORFs | none generalizable; requires protein quantification assay | disrupts inhibitory secondary structures in RNA, or blocks inhibitory uORFs |
As the table makes clear, different mechanisms can be achieved by targeting different sites within the RNA molecule to achieve different specific functions. In addition, different chemistries of ASO may permit one mechanism and prohibit another, for example, some chemistries are not RNAse H-cleavable. My next post in this series will review the different chemistries of ASOs.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.