Is there anything special about PrP's expression?
In a recent post I discussed proofs of principle for using small molecules to target one protein’s expression level. One take-home from that post is that if you want to target your protein of interest with any specificity, it is useful to find something unique about that protein, or its transcript or gene. Accordingly, this post will review what is known about PrP’s journey from the genome to the ribosome, and whether there might be any opportunities for specific intervention along the way.
chromatin state
We know of lots of small molecules that affect histone marks and therefore chromatin state: HDAC inhibitors, BET bromodomain inhibitors, and so on. So it’s not implausible to imagine that a drug modulating histone marks could affect PRNP expression.
But to imagine any kind of specificity against PRNP is much harder. In fact, I am not aware of any really good datasets available for asking whether there is anything very unique about the chromatin surrounding PRNP. If you look in UCSC Genome Browser at all of the histone mark data from ENCODE and other genome-wide projects, you are confronted with the fact that all the ChIP-seq experiments are done with antibodies against the most common histone marks, which by definition will never be specific to one gene. Here is a screenshot of this UCSC view:

All you see is a bunch of H3K4 methylation around the promoter and first exon, a smattering of H3K36 methylation throughout, and a relative dearth of H3K27 methylation — pretty much like any active gene. New, more exotic histone marks are discovered from time to time, as mentioned in molecular biology 15, but I’m not aware of a lot of genome-wide data for these. I’m probably just uneducated on the subject (I don’t follow epigenetics much), so there may well be a better browser out there somewhere with more comprehensive data (feel free to let me know as much in a comment).
transcription initiation
A couple of years ago I wrote a post reviewing what’s known about PrP transcription. While there are several reports in the literature, not much has been reproduced by different independent investigators. According to ENCODE, the PrP promoter is bound by STAT3, AP-1, and AP-2. Literature has also pointed to p53 and Sp1 as being involved with the promoter. These are all factors that regulate tons of genes. A few different investigators have reported that copper promotes, or is required for, PrP expression, but as reviewed here, people have already tested copper chelators as therapeutics for prion disease in vivo and have not shown any strong reproducible effect on disease.
splicing
I recently examined PRNP mRNA splicing in detail. Here’s a quick summary.
PRNP has only two exons and one intron, thus one splice donor and one splice acceptor. The only interesting thing about its splicing is that the donor undergoes alternative splicing, with most of the copies of the mRNA being spliced at a canonical donor motif with GT at the +1 and +2 positions, but somewhere around 30-50% of copies being spliced at a GC motif a few bases upstream. These alternate donor sites are annotated in UCSC:
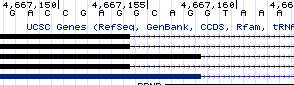
And I was able to find evidence for them in RNA-seq data as well. This is at least a little bit unique, with <1% of splice sites genome-wide having GC donors, but since no more than half of copies of the mRNA are spliced in this way, it doesn’t seem like the most compelling target.
mRNA stability
As discussed in a recent post, there is really no good proof of concept out there for it even being feasible to specifically bind an mRNA with a small molecule. Nevertheless, I figured it was at least worth looking into whether there is anything special about PRNP mRNA.
There have been two published papers [Wills 1992, Barrette 2001] asserting that PRNP mRNA forms a pseudoknot, which refers to a particular structure where RNA loops back on itself. These two papers mostly seem to have been written to propose RNA-related alternative explanations for the etiology of prion disease, which are no longer relevant given the overwhelming evidence that prions are formed by post-translational conformational change. In any event, neither of these papers includes any experimental evidence, only predicted structures. And even if the “prion pseudoknot” is real, that doesn’t necessarily imply that PRNP mRNA is unique and therefore targetable, as pseudoknots are “among the most prevalent RNA structures” [Staple & Butcher 2005].
I grabbed the whole human PRNP transcript (RefSeq variant 1, NM_000311.3) from Genbank and tried to feed it into a modern mRNA structure prediction tool called SHAPE, which apparently unlike older tools can actually handle pseudoknots [Hajdin 2013]. Sadly, it is limited to 2500 bp, while NM_000311.3 is 2755 bp, so I had to lop off the last 255 bases, which arguably defeats the whole point. Anyway, it returned to me a set of 20 PDFs of different predicted mRNA structures; here is one example [PDF]:
The other 19 looked, to my untrained eye, about the same, and I feel I have no ability to evaluate which of them is most likely or whether any of them contains a structure that is unique in the world of human mRNAs. There is no experimental evidence out there for what PRNP mRNA actually looks like, and I have trouble imagining a screening strategy that would let you specifically identify compounds that bind (and destabilize) only PRNP mRNA and no other mRNAs.
translation initiation
One potentially interesting thing about PRNP translation is its reported regulation by the presence of multiple upstream open reading frames (uORFs) [Moreno 2012]. I have touched on this topic twice briefly, in biological literature 09 and in molecular biology 27. A minority of mRNAs, the canonical example of which is ATF4, have the unique property of being translationally upregulated under conditions of cellular stress at precisely the moment when all other mRNAs are having their translation downregulated, thanks to uORFs. The principle at work here is diagramemd in Figure 4 of [Jackson 2010] or Figure 3 of [Spriggs 2010]. Basically, when the concentration of translation-ready eIF2 ternary complexes is high (normal conditions), then ribosomes acquire them quickly while scanning the mRNA, and so an upstream ORF will be translated, precluding the translation of the canonical ORF. Only when that concentration decreases (stress conditions, resulting in PERK phosphorylation) does it take longer, on average, for the ribosome to pick up such a ternary complex, such that the upstream ORF gets missed and the canonical ORF is translated instead. This regulatory mechanism certainly seems counterintuitive, but according to the reviews above, it’s well-established for ATF4.
The evidence for this mechanism applying to PRNP in particular is reported in Figure 2J and Figures S4-5 of [Moreno 2012] and is based on polysome fractioning. Under stress conditions, PRNP mRNA shifts towards a heavier fraction, suggesting association with a greater number of ribosomes, and thus implying a greater translation rate. The shift illustrated here is rather subtle. So I’d really like to see some ribosome profiling data, hypothetically demonstrating that the abundance of ribosomes on the start codon of PRNP’s canonical ORF is increased under stress, before I’d feel certain that I believe that this mechanism really applies to PRNP. I say that, particularly because PRNP does not fit the canonical arrangement of uORFs depicted in Figure 4 of [Jackson 2010] or Figure 3 of [Spriggs 2010]. According to those models, the last ORF before the canonical one should be in a different frame than the canonical ORF, and should overlap the canonical ORF, such that translation of the former precludes translation of the latter. In human PRNP, there are four uORFs, the last of which actually in the same frame as the fifth and canonical ORF, and ends long before the canonical ORF begins:
import re # python library for regular expressions
human_prnp = 'ATTAAAGATGATTTTTACAGTCAATGAGCCACGTCAGGGAGCGATGGCACCCGCAGGCGGTATCAACTGATGCAAGTGTTCAAGCGAATCTCAACTCGTTTTTTCCGGTGACTCATTCCCGGCCCTGCTTGGCAGCGCTGCACCCTTTAACTTAAACCTCGGCCGGCCGCCCGCCGGGGGCACAGAGTGTGCGCCGGGCCGCGCGGCAATTGGTCCCCGCGCCGACCTCCGCCCGCGAGCGCCGCCGCTTCCCTTCCCCGCCCCGCGTCCCTCCCCCTCGGCCCCGCGCGTCGCCTGTCCTCCGAGCCAGTCGCTGACAGCCGCGGCGCCGCGAGCTTCTCCTCTCCTCACGACCGAGGCAGAGCAGTCATTATGGCGAAC' # human PRNP sequence up just past canonical ATG, from http://www.ncbi.nlm.nih.gov/nuccore/NM_000311.3
[m.start() % 3 for m in re.finditer('ATG',human_prnp)] # calculate the reading frame of each ATG
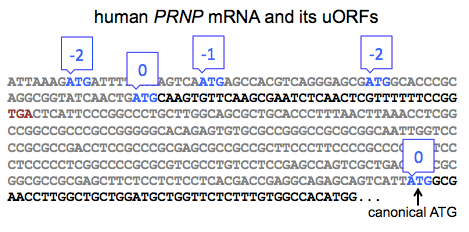
Based on this, it is not immediately obvious that the same mechanism from ATF4 should apply. Indeed, this is implicitly acknowledged in Figure S6 of [Moreno 2012], which shows the reading frames for all of the human PRNP uORFs, none of which overlap the canonical ORF. To me, this further raises the bar for the level of empirical evidence that would be required to demonstrate that an ATF4-like mechanism applies to PRNP, and I don’t think that’s a bar that’s been met yet, though I’d love to see more data on it.
In any event, supposing for a moment that PrP translation is indeed regulated by this mechanism, how would you go about trying to find a compound to interfere with it and downregulate PrP? As far as I can tell from those reviews [Spriggs 2010, Jackson 2010] there are no special proteins involved in this mechanism, just changes in concentrations of essential translation initiation factors. So a drug target doesn’t obviously present itself here. The unbiased way to search for a compound to interfere with PrP’s translation initiation would be simply to screen for compounds that reduce the activity of the PRNP 5’UTR. Say, maybe by fusing that UTR to luciferase and screening for compounds that reduce luciferase activity. That’s exactly what Jack Rogers and the folks at Broad’s Center for the Development of Therapeutics did years ago. They screened the entire MLPCN compound library at the time, 335,011 compounds, and deposited the data in PubChem as BioAssay 488864, without ever publishing the results. For a deep dive on whether there might be anything promising in there, see my blog post on PrP knockdown screens.
co- and post-translational events
I originally envisioned this post’s scope would be “genome to ribosome” but when I discussed some of these topics and those of my related post in a brainstorm session, a few of my Broad Institute colleagues tossed around a some other possibilities further downstream.
First, PrP has a signal peptide that directs it to be co-translationally translocated into the ER. Signal peptides are a fairly diverse bunch [Martoglio & Dobberstein 1998]; could one imagine a small molecule that specifically binds to PrP’s signal peptide? I went to UniProt and ran a BLAST search for human PrP’s signal peptide, MANLGCWMLVLFVATWSDLGLC, and all 250 out of the 250 results are just orthologs of PrP in other species, even when you get down as low as 63% identity. So yes, PrP’s signal peptide is pretty different from any other signal peptide in all the accumulated proteomes of UniProt. What would you do if you could bind the signal peptide with a small molecule? You wouldn’t want to just shunt PrP translation into the cytosol, because ectopic cytosolic expression of PrP is also a bad thing [Ma 2002, Ma & Lindquist 2002] (there are lots of ways PrP can go wrong). The useful thing would be if you could somehow gum up the whole complex of the ribosome, the PRNP mRNA, the signal peptide, and the signal recognition particle, in a way that keeps PrP from ever being translated. Maybe then you’d be playing with fire — it seems like there’d be a risk that anything you find would just inactivate the whole ribosome, and thus be very toxic. But who knows, the idea might be worth exploring.
The other topic that came up was PrP’s GPI anchor. One colleague pointed out that there are only about 45 GPI-anchored proteins in humans [citation needed], so PrP’s GPI anchor is a relatively unique thing. I couldn’t find a citation to support the specific number 45, but this table only lists 14 human GPI-anchored proteins, and all three reviews of GPI anchors that I looked at [Englund 1993, Paulick & Bertozzi 2008, Ferguson 2009] actually mention PrP itself, so it’s certainly one of a small cohort. There are humans who have a GPI biosynthesis defect due to a recessive mutation in the promoter of PIGM [Almeida 2006, OMIM #610293]. This results in a pretty severe Mendelian disease, which I take as a sign we shouldn’t go tinkering with GPI anchors just to get at PrP. In addition, analogous to the problem with cytosolic PrP in the last paragraph, secreted PrP is also bad, so it wouldn’t be enough to just prevent the GPI attachment of PrP — you’d have to somehow leverage targeting the GPI anchor to block production of PrP entirely.
conclusions
Here’s a quick summary of what I think I’ve learned in writing this post. I don’t see an obvious path forward for reducing PrP expression with a small molecule. One could imagine going after histone modifications or transcription factors but it would be hard to achieve specificity, and we don’t really know of anything very unique about PRNP in those departments as of now, though there could be something out there. Targeting that non-canonical GC splice donor seems like one unique thing, and the proof of principle for targeting splicing with a small molecule is certainly better than for many other mechanisms (as discussed here), but that non-canonical splice doesn’t even account for half of copies of PRNP mRNA. PrP’s mRNA might contain some unique structures, but we have neither any empirical data on this, nor any proof of principle for specifically binding one mRNA with a small molecule. PrP is reported to have a relatively rare mechanism of translation initiation regulation based on upstream open reading frames, but the evidence for this isn’t rock solid. PrP’s signal peptide may be unique, but it’s small and you’d have to find a small molecule that did just the right thing to it, blocking translation without just shunting PrP into the cytosol. And GPI anchors, though relatively rare, are essential. So all told, I don’t see any smoking guns here, no obvious path forward.
That still leaves the possibility of unbiased screens for molecules that reduce PrP expression, since there could well be some mechanism we haven’t thought of. Two such screens have already been published [Karapetyan & Sferrazza 2013, Silber 2014] and two others left to age in the cellars of PubChem (BioAssay 488864 & BioAssay 720640), without any new mechanisms, nor any safe and effective leads, emerging thus far. For a deeper dive on what those screens might hold, see this post. Unbiased screens like these are always hazardous in that there are presumably far more ways to downregulate PrP non-specifically (like cycloheximide!) than specifically. Part of the motivation for thinking about specific mechanisms that might target PrP is that if you knew what mechanism you were looking for, you could design just the right counterscreen to toss out all that chaff. Absent that, we’re left to do a whole lot of sifting.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.