Not every drug has its target

There are many approved drugs and even more molecular probes for which the mechanism of action (MoA) is unknown or at least controversial. Identifying the MoA of a biologically active compound is one of the principal challenges in phenotypic drug discovery [Vincent 2022]. Yet people often assume that the MoA boils down to one target, one specific biomolecule that can be expressed as a UniProt ID or a gene symbol. That may not always be the case. My goal in this post is to give several examples of drugs or probes whose activity may not be mediated by binding to any specific macromolecule.
This is a subject I approach with a lot of humility. It is amazing just how hard it can be to confidently determine a drug’s mechanism of action — even after well over a century in the case of general anesthetics. Add to that, none of this is even my area of expertise, so whatever level of uncertainty exists in the minds of scientists who are actually experts in this stuff, there’s yet a lot more uncertainty in my mind, trying to read enough papers to piece together what’s accepted and what’s refuted, what’s known and what’s mysterious. But I wanted to try to write this post anyway, because I think this topic is so important. If some molecules can be bioactive without even binding a target, then this opens up a whole new space of possible answers to the question “how does this molecule work”, and that’s a space I suspect we may need to consider more often than we do.
Here are the examples that I think I found.
zinc oxide: reflector
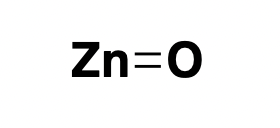

Above: a mineral sunscreen with active ingredients titanium dioxide and zinc oxide.
Mineral sunscreens, such as zinc oxide, have a mechanism of action that’s dead simple: they reflect UV light [Geoffrey 2019]. They also reflect a fair amount of visible light, which is why they appear white and don’t fully rub into the skin. Perhaps for this reason, today they mostly enjoy a niche market for people with sensitive skin, thanks to their low allergenicity. The market is more dominated by so-called chemical sunscreens, which also make our list — see next item.
oxybenzone: absorber


Above: a “chemical” sunscreen with active ingredients avobenzone, homosalate, and octocrylene.
Most active ingredients in “chemical sunscreens”, such as oxybenzone, work simply by absorbing UV radiation [Latha 2013]. That’s right, it’s just a molecule that’s very opaque in UV wavelengths. Some are more opaque in UVB (280-315 nm) or UVA (315-400 nm), some are super opaque across the whole range. Would any-old opaque molecule work? Of course, besides absorbing UV light, these molecules also need to have the right pharmacokinetic properties, if you want to call them that: the ability to stick to skin even in the presence of sweat and seawater. Add to that, they shouldn’t be irritating to the skin, and people generally prefer if they are fairly transparent in the visible spectrum.
calcium carbonate: neutralizer


Above: a generic antacid formulation of calcium carbonate.
Antacids such as calcium carbonate, used for heartburn or indigestion, are simply alkaline substances that raise pH in the gut. They are sometimes called neutalizers, and some but not all have substantial buffering capacity. I call them neutralizers here to distinguish them from the class of alkalinizers discussed further below.
simethicone: surfactant

Above: a liquid formulation of simethicone for infants.
Simethicone is an over-the-counter drug that, in my personal experience raising two kids, is extremely effective at reducing fussiness in infants. It is simply a surfactant, nothing more, and you can see it work. Shake a baby bottle full of milk, add a few drops of simethicone, and poof, no more bubbles. By dispersing foam, it presumably makes babies’ stomaches feel less bloated and achy. It isn’t systemically absorbed, but does everything it needs to do right in the gut.
polyethylene glycol: osmotic


Above: a generic formulation of PEG with average molecular weight 3,350 Da.
Polyethylene glycol, or PEG, is an over-the-counter drug for constipation. It’s a large polymer and so one molecule of PEG binds, say, 100 water molecules, effectively increasing the osmolarity of the digestive tract, thus drawing more water out of the circulation and into the gut, or conversely, preventing water in the gut from being absorbed into the circulation. Like simethicone, it’s not systemically absorbed and just acts in the gut. The typical dose is massive: 17 grams.
quinine: alkalinizer


Above: cinchona bark, a natural source of quinine.
The natural product quinine and all of its synthetic analogs — quinacrine, chloroquine, mefloquine and so on — all share the mechanism of alkalinizing lysosomes. I dove into this mechanism in much greater detail in this blog post, but here’s a quick summary. These compounds are membrane-permeable as bases, but not as their conjugate acids. They can bounce around randomly around between different cellular compartments until they hit a low pH and get protonated. Once that happens, they’re stuck. As a result, they accumulate in whichever membrane-bound compartment has the lowest pH, and they soak up those protons until the pH rises [Yayon 1984, Yayon 1985]. In the malaria parasite P. falciparum, they accumulate in the vacuole, which prevents the parasite from sequestering toxic heme from its blood meals into a crystalline form. Thus, these compounds found early use as antimalarials. The chief resistance mechanism that the parasite has developed is to upregulate a transporter that kicks the compounds back out of the vacuole [Fidock 2000]. In mammalian cells, quinine and its analogs accumulate in lysosomes (they’re often described as lysosomotropic) and are sometimes used as tool compounds to block lysosomal/autophagic degradation of proteins.
DNP: ionophore

2,4-dinitrophenol, or DNP for short, is an ionophore, more specifically a protonophore, meaning it can bind protons and shuttle them across membranes to which they would otherwise be impermeable. In this sense, its mechanism of action is the inverse of quinine. Alkalinizers move to the protons, ionophores move the protons. The electron transport chain, where we get our energy, relies upon creating an electrochemical proton gradient across the mitochondrial inner membrane (see this, or my old notes for a refresher). It uses energy from our food to pump protons out of the mitochondrial matrix across the inner membrane, and then to get back in, the protons have to go through ATP synthase, thus generating ATP. DNP short-circuits that process by allowing protons to cross the membrane with impunity. Thus, food energy is converted to heat instead of ATP. In other words, the electron transport chain is “uncoupled” from ATP production, so some call these compounds “uncouplers”. This dissipation of food energy as heat is what brown fat is actually supposed to do and is thus a key part of how hibernating bears and human infants stay warm. But uncoupling by DNP is unregulated and is systemic throughout all different cells and tissues, and is thus really dangerous. DNP was used clinically as a weight loss pill in the 1930s, and is still taken illegally for the same purpose, but it’s got an incredibly narrow therapeutic window and has led to dozens of deaths [Grundlingh 2011]. Multiple times in recent years, FDA has cracked down on websites that sell it.
edaravone: scavenger

Edaravone is a free radical scavenger approved in the U.S. for ALS (though it only helps in a certain subset of patients, for reasons unknown) [Writing Group 2017] and also approved in Japan for stroke. It has been shown to scavenge various free radicals [Watanabe 1994], though exactly which ones are relevant in ALS (or stroke) was not obvious to me from a quick read of the literature, and may be an unknown at this point.
ether: lipid interactor (controversial)

Above: monument to the discovery of ether as an anesthetic, in Boston’s Public Garden.
There has been extensive study of the mechanism of action that might be shared between (diethyl) ether, newer anesthetics such as isoflurane and propofol, and probably that oldest anesthetic of all: ethanol. Plenty of evidence points to GABA receptors as ultimately mediating their activity [Davies 2003, Hemmings 2005, Garcia 2010, Pavel 2020] — but how exactly they modulate GABA receptors is less clear and, though this is controversial, it might involve an indirect mechanism.
If anesthetics directly bound GABA receptors, then you might expect they ought to have a structure-activity relationship consistent with a specific binding pocket, like benzodiazepenes do [Hadjipavlou-Litina 1994]. Instead, an early observation was the Meyer-Overton correlation, that lipophilicity is a or the major predictor of a compound’s anesthetic potency. It was long hypothesized that by interacting with the lipid bilayer of neurons, these compounds must somehow change biophysical properties of the membrane, which might in turn change how GABA receptors behave. But what, exactly, anesthetics might do to membranes may not be simple and is a subject of ongoing investigation and controversy [Herold 2017, Weinrich & Worcester 2018, Pavel 2020]. At clinically relevant concentrations, anesthetics don’t actually seem to change bulk membrane properties much [Herold 2017], though one recent report suggests they may indeed change the behavior of lipid rafts [Pavel 2020].
Admittedly, in opposition, there exists a large body of evidence arguing that anesthetics may actually directly interact with GABA receptors and other ion channels. There is a size limit on anesthetic alcohols, which is hypothesized to reflect the maximum capacity of some binding pocket [Davies 2003]. On one hand, the idea of a binding pocket may seem inconsistent with the bizarre SAR of anesthetics, although on the other hand, their effective doses are fairly high (example: a blood alcohol content of 0.1% means 1 g/L ethanol, divided by MW 46.1 g/mol, equals 22 mM), so they needn’t be tight binders. One of the strongest arguments for direct interactions with receptors is that some specific mutations in GABA receptors have been shown to confer resistance to some anesthetics [Mihic 1997]. But it’s not clear to me that this proves that the anesthetics directly bind the receptor — couldn’t that mutation also serve to abolish the receptor’s response to some kind of altered membrane condition? Moreover, some mutations have dramatic effects on the sensitivity to one anesthetic and little effect on the sensitivity to other anesthetics [Jurd 2003], which makes it appear that direct GABA receptor binding may not provide a satisfying, unifying explanation for all anesthetics.
Overall, my read of the literature — admittedly as quite an outsider — seemed to suggest a lot of residual controversy, and a possible role for a non-protein-binding MoA for anesthetics.
reflections
I suspect there are many other examples out there, and I would love if readers of this blog point me to those examples. I considered including examples such as antibiotics that target the cell wall, or chemotherapies that target DNA by intercalation or alkylation. But these do bind a biological macromolecule, even if they’re not specific to one gene or protein symbol. I also considered several examples that I suspect might have non-target-binding MoAs, but where I ultimately felt like not enough is yet known: dimethyl fumarate, guaifenesin, and lithium. To be sure, there are theories of mechanistic targets, for instance, lithium may act through GSK3β, dimethyl fumarate might acts through Nrf2 [Mills 2018], but that doesn’t necessarily mean direct binding, and sometimes not all the evidence agrees [Schulze-Topphoff 2016].
The examples I found above are generally either very small or very simple (e.g. repetitive polymers). Sometimes the dose requried is enormous. Their structure-activity relationships might provide some hint as to their unique MoA, for instance, general biophysical or biochemical properties — things like pKa, cLogP — may be more important for activity than any particular structural motif like you would see with a protein-binding MoA. But beyond that, I don’t see any recipe as to how to identify an MoA of this nature. Clearly, many of the cutting-edge techniques for identifying target proteins, like seeing if the target is pulled down by a conjugated analog [Ong 2009], gains thermal stability upon binding [Reinhard 2015], or is mutated when you select for resistance [Kato & Comer 2016] won’t be useful in these cases.
If nothing else, the above examples illustrate that there are a variety of MoAs not mediated by binding a specific biomolecular target. This is one possibility that should be considered when one is hunting for the mechanism of action of drug or probe.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.