Viral expression of antibodies in the brain as a therapeutic strategy in prion disease
Last week Sonia and I sat down for a brainstorm session with the Harvard gene therapy researcher Luk Vandenberghe, on how to best get large biomolecules into the brain to treat prion diseases. Hot off of doing much of the reading for my last post on CRISPR/Cas9, I came into the meeting most interested to talk about the potential for genome engineering to knock out PRNP once and for all. But Luk encouraged me to also consider a different strategy: using viral vectors to express antibodies in the patient’s own cells. That’s what this post will be about.
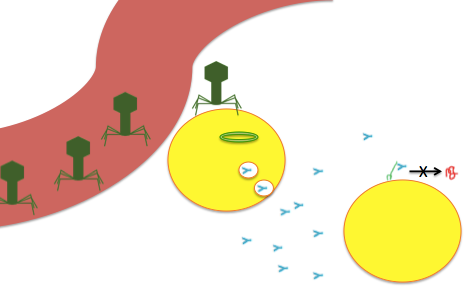 An impressionistic drawing of the viral-delivery-of-antibodies-against-PrP strategy. Viruses (drawn as bacteriophages even though that’s not what AAV looks like) arrive via (let’s say) the bloodstream, transduce nearby cells with a viral genome (depicted as circular) which encodes a secreted antibody (blue Y) which is released into the brain where it can bind PrPC (green squiggle) and prevent its conversion to PrPSc (red squiggle).
An impressionistic drawing of the viral-delivery-of-antibodies-against-PrP strategy. Viruses (drawn as bacteriophages even though that’s not what AAV looks like) arrive via (let’s say) the bloodstream, transduce nearby cells with a viral genome (depicted as circular) which encodes a secreted antibody (blue Y) which is released into the brain where it can bind PrPC (green squiggle) and prevent its conversion to PrPSc (red squiggle).The biggest motivation for putting an antibody, rather than CRISPR/Cas9 or shRNA, into a viral vector is to amplify the therapeutic effect beyond the few cells you can directly reach with your viral vector. At present, AAV9 seems to be the state of the art in terms of viral vectors, being relatively low-immunogenic while also achieving higher expression levels than other AAVs across many tissues, including the brain, following intravenous injection [Simonato 2013, Zincarelli 2008 (ft)], and also does well after intraventricular injection [Bucher 2014 (ft)]. In small animals, some real successes with AAV9 have been reported [Lee 2014]. However, even with AAV9, distribution in the CNS is fairly limited. In rodents, the virus has been reported to reach more than 50% of neurons in the spinal cord, Purkinje cells of the cerebellum and a few other choice brain regions [Foust 2009]. But when one considers the whole brain, particularly in larger animals such as pigs, dogs, and non-human primates, it can be difficult to find hard numbers [Bevan 2011]. One study using intrathecal injection found that AAV9 reached just 2% of the whole brain in non-human primates [Gray 2013].
One way to amplify your impact is to take the small percentage of cells you can directly reach, and turn them into factories to produce a secreted protein that can in turn circulate more widely throughout the brain. There are a few different types of proteins one might want to deliver. For instance, in animal models of loss-of-function lysosomal storage diseases, people have used AAV9 to transduce epithelial cells in the brain and have them secrete enzymes to replace those lost due to the genetic mutations. For instance, enzymes like β-glucuronidase (human gene symbol: GUSB) or tripeptidyl tripeptidase I (TPP1) – these proteins are normally found in the lysosomes, so if you can secrete them and then have them be endocytosed by deficient cells, you can potentially get into the correct subcellular compartment. One neat study used a phage display library to select for peptides to display on the surface of virus caspids in order to get the virus taken up into brain epithelial cells so that those cells could secrete these enzymes to be taken up by neurons [Chen 2009]. That study saw some effect on disease phenotypes and managed to raise TPP1 levels in a couple of particular brain regions up to the levels in het knockouts. Though prion diseases are caused by a gain of function, the closest analogue of this approach might be to deliver dominant negative versions of PrP itself to interfere with prion propagation, and a couple of studies have looked at this therapeutic route [Crozet 2004, Furuya 2006] though I won’t go into detail on that here.
Another approach, which has gained currency in the HIV field, is to use viral vectors to deliver antibodies. In the case of HIV, the goal seems to be to deliver broadly neutralizing antibodies – those that work against multiple HIV strains – as a preventative measure. Apparently new HIV infections, at least in the case of vaginal intercourse, start with a very small number of “founder” viruses, so it’s (relatively) easy to stop the infection at this early stage using antibodies. Doing so with antibodies delivered in a viral vector is called “vectored immunoprophylaxis.” One recent study injected AAVs encoding a broadly neutralizing antibody intramuscularly in humanized mice, and found that this was sufficient to get the antibody circulating both in the blood and in the vaginal mucosa, and prevented HIV infection under most conditions [Balazs 2014]. This approach could be a valuable prophylactic in humans: it’s easier to get a viral injection to people once than an antibody injection to them biweekly for the rest of time. And for what it’s worth, antibodies are really expensive to manufacture, so having people’s own cells produce them is a cost-saver too.
Of course, blood and mucous membranes are the “easy” tissues to reach, compared to the brain. If you can only reach a small fraction of cells in the brain, you’ll want to really highly express the antibodies in order to maximize their chances of reaching the cells that need them.
This approach is appealing in prion disease because there exists at least some proof of principle to show that antibodies can clear or prevent prion infection. An ideal proof of principle might be to make a mouse that constitutively expresses an antibody, and show that this mouse cannot be intracerebrally infected with prions. Better still would be to make a mouse that conditionally expresses an antibody, and then to turn on expression after much of the prion disease incubation time has passed and symptoms are beginning to show, and see if the antibody can reverse the experiment. Instead, what has been published is that constitutive expression of the 6H4 or 15B3 antibodies can prevent peripheral prion infection [Heppner 2001]. The 15B3 antibody is specific to PrPSc [Korth 1997], and I couldn’t find a Kd value published anywhere, though this exchange implies its affinity is much poorer than some of the antibodies out there for PrPC, including 6H4. Which brings us to the second proof of principle: that intravenous injections of antibodies can also clear peripheral prion infection if given before prions reach the brain [White 2003]. That study used ICSM-18 and ISCM-35, the former of which has excellent affinity for PrPC: Kd = 0.13 nM [Antonyuk 2009 (Supp Table S1)] and has since been humanized as PRN100.
The White study also tried IV injections of antibodies in intracerebrally infected mice as well as in peripherally infected mice after neuroinvasion, and found no efficacy in either model. A pharmacokinetic explanation is sufficient to explain this: antibodies in the bloodstream just don’t usually cross the blood brain barrier in appreciable quantities – at least, not without a “brain shuttle” or something fancy. Direct infusion of antibody into the brain after intracerebral infection has achieved far more modest effects, extending survival time by just 8% [Song 2008 (ft)]. The more limited efficacy in that study is surely at least partly because the infusion could only be done for a limited time, and even with direct infusion it is difficult to disperse antibodies throughout the brain.
Still, though pharmacokinetic explanations are sufficient to explain these shortcomings, we are left without any solid proof of principle that antibodies in the brain can prevent or reverse prion disease. The experiment of intracerebrally infecting a transgenic mouse constitutively expressing anti-PrP antibodies seems far too obvious for no one to have done it by now, though I suppose that waiting 600 days to show that a mouse doesn’t get sick is an unrewarding way to spend one’s time.
Supposing for a moment that pharmacokinetics is the only barrier here, the idea of using a virus to transduce a few epithelial cells or neurons or glia throughout the brain to produce antibodies that will reach their neighboring neurons seems like a promising way to get better coverage of the brain.
From my search of the literature, it looks like three studies have attempted this approach in prion disease [Wuertzer 2008, Zuber 2008, Moda 2012].
The first study demonstrated modest but clear therapeutic efficacy with this approach [Wuertzer 2008]. Full-length antibodies as well as Fabs (antibody fragments) require multiple proteins encoded in constructs that end up being on the large side for viral vectors, so this study instead used single chain variant fragment (scFv) antibodies, where the heavy and light chains are fused via a linker region into a single peptide. The researchers constructed an scFv version of the well-characterized D18 antibody [Peretz 2001] and also screened phage display libraries to discover a few novel scFvs that bound PrP. As a negative control, they used an scFv that binds phenobarbital, and has no affinity for PrP. They used an AAV2 vector, and performed four intraparenchymal (direct into the brain tissue) injections per mouse, in the left and right striatum and thalamus, with about 1e10 “expression units” ( = viral genomes?) per injection [supplement]. They found evidence (using AAV expressing GFP) that the virus reached places in the thalamus up to about half a millimeter away from the injection site [supplement S3] and that the secreted scFvs traveled even a bit further afield, being found in the hypothalamus. So: not a ton of coverage of the whole brain, but it does appear that they were able to amplify their reach using the secreted protein strategy. The AAV injections were done prophylactically – a whole month ahead of time – and the scrapie inoculations were done peripherally. The mice treated with the scFv D18 vector had disease onset at 250 days post-infection (dpi) while controls had onset at 199 days. The ranges of disease onset times between control and treated were non-overlapping and the result was significant (p = .013, log-rank test) even after Bonferroni correction. As an aside, the authors have earned some serious respect from me by being the only ones I’ve seen to actually apply multiple testing correction to a mouse therapeutic study.
That study provides a good proof of principle. The clearly significant 25% delay in disease onset establishes that this approach can have some effect in prion disease, and remains to this day the best outcome achieved with this approach. That said, the study design is a long ways off from anything that would mimic the situation in the clinic. Readers of this blog will know I’m generally not fond of experiments where mice are inoculated peripherally, nor of prophylactic experiments. Human cases of peripherally acquired prion disease are vanishingly rare, necessarily unplanned, and in any event go unnoticed until neuroinvasion occurs and symptoms present. Humans are only available for treatment once the prion infection is in their brain and most of the incubation period has passed.
The same year, a study by a different group took a similar approach but using antibodies against the 37 kDa laminin receptor (LRP for short; human gene symbol: RPSA) instead of against PrP itself [Zuber 2008 (ft)]. The motivation for this was that group’s lengthy body of work on LRP as a receptor for PrPC and PrPSc. This focus began with the discovery that PrP binds LRP and that LRP levels change during prion disease [Rieger 1997], the rest of the saga is briefly reviewed in [Zuber 2008 (ft)]. It is not clear to me if LRP has ever been established as actually being necessary for prion infection. From Googling it seems that someone has made RPSA knockout ES cells, but I couldn’t find a paper where a knockout mouse has been characterized (if you know otherwise, leave a comment below). Possibly that’s because it’s embryonic lethal but I couldn’t find a paper reporting that either. They generated a transgenic mouse expressing siRNA against LRP, and characterized it [Leucht 2004] but as far as I can tell never reported the effect on scrapie incubation time other than to state that this siRNA “delays the incubation time in scrapie-infected mice (H. Ludewigs and others, unpublished data)” [Zuber 2008 (ft)].
In any event, these researchers put an anti-LRP scFv into an AAV2 vector and microinjected 5e9 “genomic particles” (as above, I assume this is the same as viral genomes or vg for short) into the hippocampus. This was done prophylactically (14 days before infection), and the mice were infected intracerebrally. The authors reported a reduction in PrPSc in the spleen at 90 dpi, but the effects on brain PrPSc were not characterized – odd, since this was with intracerebral injection. There was no effect on time to disease onset or death [Table 1].
I’ll briefly mention one other approach: one study expressed an scFv version of the 3S9 antibody against PrP ex vivo in a microglial cell line and then injected those cells prophylactically into mice before prion infection [Fujita 2011]. This approach had a very slight effect – it looks like the average survival of treated mice was about 175 dpi compared to 170 in controls, though apparently it was significant (p < .02).
Fast forwarding, the most recent effort used an scFv version of the D18 antibody, delivered via an AAV9 vector [Moda 2012]. Once again, the treatment was prophylactic (though the paper doesn’t seem to say how many days in advance of prion infection), by direct intraparenchymal injection into the brain (hippocampus, hypothalamus and thalamus, all on the right side). They first used a β-gal assay to visually examine the distribution of AAV9 in the brain – it was better than you might expect based on just three injection sites, but by no means comprehensive coverage [Figure 1]. The study found evidence of reduced PrPSc burden in treated mice both at intermediate and terminal stages, and the time of onset based on behavioral tests was delayed by 13% (187 dpi vs. 166 dpi, p = .02) but the difference in survival time was merely suggestive (202 dpi vs. 188 dpi, a 7% delay at p = .09).
So that seems to be the progress so far: even with prophylactic treatment against peripheral infection – far from the most stringent test of efficacy – antibodies delivered by viral vectors have barely been able to move the needle on prion incubation times. And that’s in mice, whose brains are less than a thousandth the size of ours.
It is interesting to brainstorm what could be done to improve upon the efforts that have already been invested in this approach. Perhaps a better antibody would make a difference: ISCM18 has ten-fold stronger affinity for PrP than D18 does, with a Kd of .13 nM [Antonyuk 2009 (Supp Table S1)], compared to 1.6 nM for D18 [Peretz 2001 supplement], so if this could be maintained in scFv form that would be compelling. As an added bonus, the humanized version of ICSM18, called PRN100, may also see a clinical trial by IV injection of antibody, in which case the safety of the antibody itself will already be established, leaving one fewer obstacle in the way of bringing vectored antibodies to patients.
It would also be interesting to see whether intravenous delivery could achieve a broader distribution of antibody than the intraparenchymal direct injections used in these studies. Consider that the whole point of the vectored antibody approach is to hijack the cells you can directly reach with a vector and get them to indirectly reach their neighbors with an antibody. Reaching, say, 5% of neurons spread evenly throughout the brain could therefore be more valuable than reaching 5% of neurons all in the hypothalamus. Admittedly, my hypothesizing that this would be better pre-supposes that the existing studies did saturate the local viral injection site with antibody, while failing to reach the rest of the brain, but that may not even be true. Moda’s Figure 5 suggests that even in the hippocampus, which received direct injection, vacuolation at the time of terminal disease was just as bad as in control mice.
Still, the possibility of using IV injection to achieve a more even antibody distribution is tempting. Of course, IV delivery has its own problems – lab mice with naive immune systems might do just fine with AAV9, but about a third of humans are seropositive for AAV9, meaning they have neutralizing antibodies that would prevent the vector from reaching the brain. The vascular system is also an inefficient way to deliver the vector because many viral genomes will be lost to peripheral tissues. But at the end of the day, the size of the vascular system in the brain scales with the size of the brain; the size of the area reached by intraparenchymal injection does not. If we want experiments to scale from mice to humans, that’s an important consideration.
Zooming out a bit, there are a number of ideas out there for improving delivery to the brain more generally: using more neurotropic viruses such as HSV-1, engineering better viruses (including the phage display strategies mentioned above), borrowing protein fragments from highly pathogenic but highly neurotropic viruses like rabies, or engineering a virus that is just replication-competent enough to reach its target without destroying the brain in the process. These strategies all have their pros and cons which I don’t know enough about yet to go into detail on in this post. But if you, the reader, have specific thoughts on how the vectored antibody delivery approach in prion disease could be improved, I encourage you to leave a comment below.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.