Prion2014 Day 3

Note: Prion2014 has no formal policy on social media, so I have been limiting my blogging to published work that speakers have discussed, except where speakers have given me permission to mention unpublished portions of their presentations.
beginning
I arrived late and missed the better part of the talks by Patrik Brundin and Michel Goedert.
David Sanders
Sanders is from the Mark Diamond lab at Washington University in St. Louis. He presented work published last week on the faithful propagation of tau prion strains in cell culture and in mice [Sanders & Kaufman 2014]. He first cited evidence that tau aggregates enter cells via macropinocytosis [Holmes 2013, Frost 2009?]. It has been suggested that such cell-to-cell transport of tau aggregates only occurs at synapses. He set out to test whether tau strains propagate faithfully in cell culture.
In the new study, Sanders used monoclonal tau RD-YFP cells, transduced them with Tau fibrils, isolated clones and then ran assays to characterize the Tau prion strains. His talk focused on three clones they isolated: clone 1 had only soluble tau, clone 9 had a common tau strain where staining revealed neurofibrillary tangles, and clone 10 had a rare tau strain where the staining had more of a punctate pattern. The staining pattern of tau was markedly different in these cells, and propagated faithfully over many passages over 6 months’ time. On Western blots, Clone 1 had no pronase-resistant material, Clone 9 had a smear of pronase-resistant material of varying molecular weights, and clone 10 had distinct bands.
They next took cell lysate from clones 9 and 10, boiled it, and used it to template new cells. They obtained multiple clones exposed to clone 9 lysate which had clone 9′s tau staining patter, and similarly for clone 10. They next used a split luciferase system, in which they transfected with constructs of tau fused to either the C-terminal part or N-terminal part of luciferase, such that only when tau aggregates is luciferase complete and able to produce light on exposure to luciferin. They also used several other assays to characterize strain differences.
They then inoculated lysates from clones 1, 9 and 10 into Tg mice expressing Tau P301S and collected brains for histology at 21 days post-inoculation. They did indeed obtain different staining patterns in mice inoculated with clone 9 vs. clone 10 lysate. They did serial passages in mice, immunoprecipitated tau and then re-introduced the strains to cell culture. By and large, they saw faithful propagation here too – the one exception was a cell exposed to clone 10 tau which had a different staining pattern; they believe this represented the tau prions spontaneously generated in vivo in the P301S mice.
They then obtained brains from 29 patients with 5 different tauopathies and introduced those strains to cells. Some brains had no seeding activity, and others diverged into five different patterns. The staining patterns largely clustered according to the disease the patients had been diagnosed with – for exmaple, Alzheimer’s patients alnmost always had the “speckled” pattern. He also praised another group’s related efforts in characterizing human Tau strains by inoculation into mice [Clavaguera 2013].
Neil Cashman
Cashman opened with a general review of misfolded proteins in several diseases and his own efforts to develop therapeutic antibodies for them. He then moved on to his main topic, which came as a surprise, to me at least: targeting PrP to treat cancer. He introduced a conformation-dependent PrP monoclonal antibody called AMF-1c-120 which he is investigating as a potential cancer therapeutic. This work has not been officially published, but as far as I can tell, all the information in Cashman’s talk can be found online. He is founder, Chief Scientific Officer and a member of the Board of Directors of Amorfix, the company which owns the antibody (a conflict which he disclosed). Their full efficacy data is contained in U.S. Patent Application WO2013185215A1, and Amorfix also offers a concise summary on their website. Cashman also presented the antibody at a cancer conference last year.
Cashman suggested that there are many reasons – pH, oxidative stress, and so on – why tumors might develop misfolded proteins. And indeed, there are antibodies against EGFR which recognize wild-type EGFR on the surface of cancer cells but not normal cells, suggesting that the antibodies are conformation-dependent and the protein is misfolded in cancer [Garrett 2009]. His company therefore developed a model to predict, a priori, which regions of proteins were most likely to become exposed upon misfolding. For PrP they predicted that the YYR and YML epitopes would be more exposed in misfolding, so they raised antibodies against them, and found that the antibodies stained more darkly on ovarian cancer cell lines than on other cells. They found that PrP on these ovarian cancer cells exhibited some degree of proteinase K resistance. A collaborator found similar results with primary lung cancer cells.
They then moved on to testing therapeutic efficacy. With only the naked antibody, they saw a reduction in tumor growth rates (I didn’t catch if this was in vivo or in vitro). They then conjugated the antibody to urease (an enzyme which makes the extracellular environment more alkaline and is therefore cytotoxic), to obtain added efficacy at killing cancer cells.
Abigail Diack
Diack apparently switched time slots, she was originally scheduled yesterday. Diack’s talk was on variably protease-sensitive prionopathy (VPSPr). Her work is unpublished, but as can be seen in Abstract O.13, she tested the transmission of a few VPSPr cases to Jean Manson’s HuPrP knock-in mice [Bishop 2006] humanized mice with 129MM, MV or VV genotypes. None of the inoculated mice had evidence of clinical signs or vacuolation. Some mice did have abnormal PrP deposits, and just one mouse had a deposition pattern that mimicked that seen in human VPSPr patients, dubbed “microplaques.” She also compared the observed transmission properties of VPSPr to those of various CJD strains as characterized in [Bishop 2010].
In the Q&A, Pierluigi Gambetti took the podium to discuss tau pathology in VPSPr. He said his group now has ~50 cases of VPSPr and most (60-70%) of them have focal tau pathology.
Federico Benetti
Benetti reviewed some of the evidence for how PrPC and copper might regulate the NMDA receptor (NMDAR), and for the S-nitrosylation of NMDAR cysteines. He hypothesized that PrPC has a neuroprotective role by copper-dependently assisting the S-nitrosylation of NMDAR. He found that S-nitrosylated GluN1 and GluN2 subunits were reduced by about half in PrP knockout mice compared to wild-type mice, and that knockout mouse hippocampal neuronal cultures were more susceptiible to NMDAR-mediated toxicity. To determine whether copper ions were required for this phenotype, he chelated copper with cuprizone and found that this treatment reduced the levels of S-nitrosylation in wild-type hippocampal cultures to the levels seen in knockout mouse hippocampal cultures. Similarly, copper chelation increased the vulnerability of wild-type cultures to NMDAR toxicity but did not affect toxicity in the knockout cultures. He showed a large diagram for the pathway that he hypothesizes mediates the NMDAR toxicity that is modulated by PrPC.
Fabrizio Chiti
Chiti presented on the structural determinants of the toxicity of protein oligomers. As a motivation for this line of research, he argued that oligomers of any given protein are surely structurally diverse, but that there might be structural elements associated with their toxicity. He discussed a few findings related to Aβ and amylin (IAPP), but most of his talk focused on the HypF-N protein, which is a focus of his lab [Campioni 2010, Saridaki 2012, Campioni 2012].
HypF-N is capable of forming two types of oligomers, dubbed Type A and Type B, which cannot be morphologically distinguished at present but are operationally defined such that A is cytotoxic when added to the extracellular medium of cells, and B is non-toxic. Type A oligomers also induce an increase in intracellular Ca2+ and reactive oxygen species (ROS), inhibit LTP in hippocampal slices and cause cholinergic neuron loss and cognitive impairment when injected into rats’ brains.
Chiti used some technique involving excimers to determine that the non-toxic B form is structured and has its hydrophobic residues buried, while A has some of the hydrophobic parts exposed and unstructured [Campioni 2010].
Chiti next determined which chaperones and which co-factors do and do not reduce the toxicity of Type A oligomers of HypF-N. As Invited Abstract #10 indicates, he found that toxicity depended upon the presence of GM1 in the cell membrane. GM1 is a ganglioside, which means a sialyated glycosphingolipid. There were some more details here too but in a quick PubMed search they don’t seem to be published yet, so I won’t go into further detail here.
Jesus Requena
As motivation, Requena argued that once we know the structure of PrPSc we will immediately understand the mechanism of prion replication, analogous to how when Watson and Crick discovered the structure of DNA, it “did not escape their notice” that the structure provided a mechanism for replication.
To elucidate the structure, Requena has focused on negative stain transmission electron microscopy of anchorless PrPSc fibrils, since they are unglycosylated and therefore relatively pure protein. His overall conclusion is that the fibrils each consist of 2 intertwined protofilaments, each of which has axially stacked β-strands repeating with a periodicity of 4.7 Å.
His TEM images show a 4.7 Å repeating unit, which should represent one beta strand. Most of the fibrils are composed of two protofibrils. Using helical reconstruction on the TEM images they created an image of the structure which shows grooves along the spine of the fibrils, and shows a hint of the latitudinal beta strands at 4.8 Å intervals.
Based on the mass of a PrP molecule, one anchorless PrPSc molecule has to occupy a volume of about 20247 Å3, so if it’s a cylinder of diameter 4.9 Å, then the height has to be ~15.3Å. This is strongly consistent with Holger Wille’s results from X-ray diffraction. By averaging TEM images of many different fibrils, they determined that the fibrils have ~4 nm axial “blobs” which must represent PrP dimers. This is consistent with Fourier transform analysis.
He suggests that prion strains represent differences in surface topographies of the upper and lower rungs.
He noted the previous finding that, using limited PK digestion and mass spec or a C-terminal antibody on a Western blot, you can identify multiple C-terminal fragments of differing sizes [Vazquez-Fernandez 2012]. They believe that the region from 151/152 to 232 is the most highly protease-resistant core.
Based on all these different sources of information, he presented a model, which he disclaimed as being highly speculative, of what the structure might look like.
In the Q&A, Surachai Supattapone asked how the disulfide bond and glycan chains would fit into the structure. Requena responded that the glycans would stick outward, and the disulfide bond would “just be there.” Ilia Baskakov asked how Requena reconciles his model, which leaves no room at all for alpha helical structure, with FTIR data suggesting there is some alpha-helical content in PrPSc. Requena said that he thinks FTIR data have been “over-read” to infer that there are alpha helices and that those peaks in FTIR could just represent loops or turns. In response to another question, Requena also clarified that his hypothesis is that strains differ in tertiary structure, not in quaternary structure. The asker responded that many prion diseases such as classical scrapie, atypical scrapie, and BSE do not feature fibrils at all, and therefore the quaternary structure must differ between strains.
Janez Plavec
Plavec started from the NMR structure of full-length PrPC [Zahn 2000] and asked how different genetic prion disease mutations promote misfolding and how dominant negative mutations prevent misfolding. He obtained samples of HuPrP 90-230 with the Q212P, V210I and E219K variants. He noted that there are no differences in structure of alpha helix 3 between Q212P and wild-type PrP but that when you have a “broken” alpha helix, Q212P changes the angle between the two parts [Ilc 2010]. He also discussed the reported structures of V210I PrP [Biljan 2011] and of E219K PrP [Biljan 2012]. In V210I PrP, the I210 residue must be profoundly buried, because in deuterium exchange, it has almost no exchange with deuterium even after 3 months of being dissolved in deuterated water. He concluded with some data on the pH dependence in the structures. I believe most or all of this is covered in a review he published last year [Biljan 2013].
A question-asker wanted to know if the mutations affect overall stability of PrPC. Plavec argued that the mutations have only a local effect but that this might trigger downstream structural changes. He said that we don’t have a method available to figure out the total ΔG of unfolding in these PrPs vs. wild-type PrP. Total ΔG is a sum of many contributions of intermediate folding states, and he believes that these mutations may only lower the energy for one intermediate change.
Glenn Millhauser
Millhauser used his talk to introduce a hypothesis tying together two recent findings from his group: a new structural model explaining the zinc-dependent interaction between the C and N termini of PrP [Spevacek 2013], and a new paradigm for alpha cleavage [McDonald 2014]. I’ve blogged in more detail about Millhauser’s work in this area in this post.
He briefly reviewed this published work. In [Spevacek 2013], they obtained NMR data on PrP with and without a single zinc ion bound to the four octarepeat histidines. All of the changes in structure occurred on the faces of helices 2 and 3. This appears to be due to a higher-order structure where the Zn2+-bound octarepeats “nestle in” against this face. He noted that all of the genetic prion disease mutations which affect charge, all increase charge, and his recombinant PrPs with the mouse equivalents of the D178N and E200K mutations show reduced interaction of the helix faces with the zinc-bound N terminus. In [McDonald 2014] they found that there are three alpha cleavage sites in the central region of PrP, and that zinc and copper change the relative abundance of the different types of cleavage.
Moving on, he introduced his theory from his recent review [McDonald & Millhauser 2014] arguing that the differences in toxicity of PrP deletion mutants can be explained as follows: the toxic deletions abolish all three alpha cleavage sites, while keeping the N terminus as long as possible, allowing it to retain its “executive function.” He believes that the toxicity may result from “overdrive” whereby the N terminus’s function cannot be downregulated via alpha cleavage.
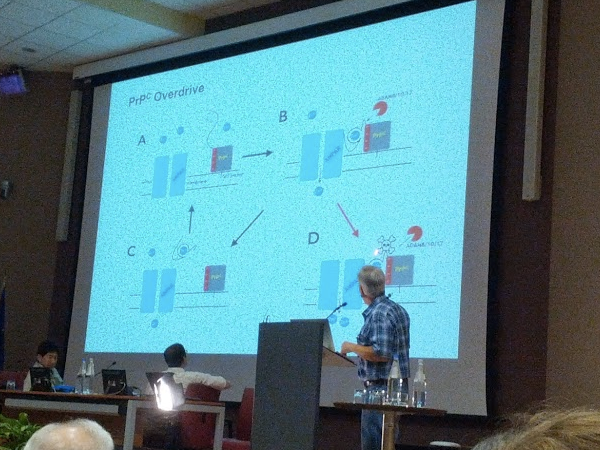
In the Q&A, Surachai Supattapone asked how Millhauser would explain the rescue of the toxic phenotype of Shmerling mutants by co-expression of PrPC. Millhauser responded that he believes it is lateral association – PrPC interacts with the mutant PrP, reducing the latter’s interaction with AMPA or another protein. Another question-asker, whose work supported the channel model of ΔCR toxicity, said his data support the octarepeat region being unimportant in channel formation. Millhauser responded that he doesn’t particularly believe the channel hypothesis, but that he thinks they can answer this question by using amino acid substitutions instead of deletions to abolish alpha cleavage. Roberto Chiesa asked whether Millhauser had looked at zinc-binding activities of PrP with octarepeat insertions. Millhauser responded that he’d only done that for copper, not zinc [Steven 2009]. Claudio Soto asked how Millhauser’s model relates to the abundance of alpha and beta cleavage in vivo. Millhauser responded that he sees ~50% of PrP being alpha cleaved in vivo, and that he believes it is protective. He thinks beta cleavage is harmful for a separate reason, which is that it leaves a lot of hydrophobic residues hanging around.
Sanjeevi Sivasankar
Sivasankar is a physicist focused on single molecule technologies who has taken a recent interest in PrP. He wanted to know how PrP misfolds and acquires proteinase K resistance, and how the misfolding of each PrP molecule changes the kinetics of aggregation into dimers and oligomers and larger aggregates.
Sivasankar used two recombinant PrP constructs: full-length recombinant MoPrP 23-231, and the globular MoPrP 90-231. He used a single molecule fluorescence assay to identify PK-resistant PrP. In this assay, PrP is immobilized on a substrate such that the distance between different PrP molecules is larger than the wavelength of light in the fluorescence readout being used, allowing you to distinguish single molecules. The immobilized proteins are then incubated with various divalent (2+) metal ions for a number of hours, and then with proteinase K. If and only if the PrP has acquired PK resistance during the metal incbuation, some PrP remain after the digestion. They then use fluorescent streptavidin to label the remaining PrP.
He found that full-length PrP exposed to copper acquires proteinase K resistance, while no other metal ions induce this, and the globular 90-231 PrP never acquires PK resistance. This has a few implications: first, it demonstrates that it is possible for single PrP molecules to acquire PK resistance, which hasn’t been shown before. Second, it shows that, at least for single molecules, this requires copper and the N terminus.
To measure the kinetics of prion aggregation, they then touch a tip coated in immobilized PrP to the surface covered in immobilized PrP, allowing the PrP to dimerize, and then pull the tip away and use an atomic force microscope to figure out how much force is required to break the PrP-PrP bond. In this assay, the globular 90-231 PrP did not bind to each other above background signal, whereas the full-length PrP 23-231 gave binding far above background. A number of different divalent metal ions were capable of increasing the probability of a PrP-PrP interaction, and copper did so most strongly. The relative on-rates and off-rates allowed them to calculate the association constant (KA). KA is a few of orders of magnitude lower – i.e. affinity is a few orders of magnitude higher – for PK-resistant PrP dimerizing than for PK-sensitive PrP dimerizing.
In Q&A, David Westaway praised Sivasankar’s work as finally clarifying that copper promotes PK resistance through an interaction with PrP and not by inhibiting PK itself, as had been previously suggested.
Chiara Zurzolo
Zurzolo’s group was the first to show, five years ago, that PrPSc can spread from cell to cell via tunneling nanotubes (TNTs) [Gousset 2009]. They have also published evidence supporting the active transport of prions from dendritic cells to neurons, suggesting TNTs could be implicated in prion neuroinvasion [Langevin 2010]. For more background, see my primer on how prions travel from cell to cell. Zurzolo’s talk focused on recent unpublished updates to this work, which I can’t share here.
Ina Vorberg
Vorberg argued that whereas PrP is GPI-anchored and so any co-factors for prion conversion must be available at the membrane, any co-factors required for cytosolic proteins would have to be found in the cytosol. The yeast Sup35 prion depends on the Hsp104 chaperone to cleave it and create new propagons [Chernoff 1995], and there is no ortholog of Hsp104 in mammalian cells, so Vorberg wondered whether it would be possible to identify a chaperone that allows Sup35 prion propagation in mammalian cells. She therefore expressed the soluble Sup35NM domain in N2a cells and seeded it with exogenous Sup35 aggregates, and found that it was indeed capable of propagating as a prion in these cells [Krammer 2009]. The prions do not form spontaneously without seeding, they can spread horizontally and also vertically on cell division [Hofmann 2013]. The aggregates do not appear to affect cell viability. More of Vorberg’s results and perspective on the relevance to neurodegenerative disease are reviewed in [Krauss & Vorberg 2013].
Peter-Christian Klöhn
Klohn presented his work, published last week, on identifying genes other than Prnp that influence prion replication in cell culture, and figuring out the functional reasons why they do so [Marbiah 2014].
As background, most cell lines cannot be infected with prions, and no one has ever figured out what is special about the few cell lines that have been stably infected with prions. Klohn started from the prion-susceptible cell line PK1 and screened for prion-resistant subclones using the Scrapie Cell Assay [Klohn 2003]. He calls these resistant subclones “revertants”, since they’ve reverted to the original prion-resistant state. He then compared microarray gene expression data from revertants and susceptible subclones. When he did a network analysis of differentially expressed genes, he found that the susceptible subclones had signatures of a greater degree of neuronal differentiation, while the revertants had less differentiated character. They therefore tested whether retinoic acid (RA), which promotes neuronal differentiation, would promote prion replication, and found that indeed it did. They then performed microarray on RA-treated and untreated cells as well, and found 18 genes that were differentially expressed in both the RA vs. no RA and susceptible vs. revertant comparisons. There were 9 genes that were downregulated in susceptible subclones, so they knocked down each of these 9 with siRNA, and found that all of them validated, in that knockdown increased susceptibility. (Of these 9, only 4 validated in CAD5 cells, a different cell line – see the Marbiah 2014 paper).
The protein products of several of these 9 genes localize to the extracellular matrix (ECM), so they then did functional work to characterize PrPSc in the ECM. They found that antibody binding to PrPSc was greatly increased in cells treated with acetone – so he believes that there is plenty of PrPSc in the ECM but that it is occluded from the reach of antibodies by cholesterol, which acetone removes. The labeled PrPSc deposits co-localize with neuronal cell adhesion molecules (NCAMs), indicating they’re in the ECM. One of the genes, Papss2, is involved in the sulfation of heparin and other glycosaminoglycans. There is a mixed literature on the role of heparin and sulfated GAGs in prion replication; Klohn now suspects that sulfated GAGs inhibit prion replication in vivo.
In the Q&A, George Carlson asked whether any of the difference in prion replication rates could be attributable to cell cycle. For instance, if one of the genes’ knockdown caused the cell cycle to slow, that would reduce the dilution of PrPSc on cell division, thus making the replication rate appear larger. Klohn acknowledged that this is a possibility, agreeing that reduced cell division rates would indeed increase PrPSc accumulation and citing [Ghaemmaghami 2007] as evidence.
Benoit Schneider
Schneider presented his work, published last fall, on the role of TACE (an alpha secretase, a.k.a. ADAM17) in prion and Alzheimer’s disease [Pietri 2013]. They found that prion-infected cerebellar granule neurons have increased TNFR1, increased PDK1 and decreased TACE activity. During prion infection, TACE relocates away from the membrane and into vesicles, making it less able to cleave PrP. They believe that prion infection leads to an excess of Src family kinase (e.g. Fyn) signaling, which increases PI3K activity, which increases PDK1 activity, which internalizes TACE. They then showed that BX912, a PDK1 inhibitor, could reverse the changes in TACE in prion-infected neurons. In 22L prion-infected mice treated ~80% of the way through the disease course (treated at 130 dpi, controls dead at 166 dpi), this compound extended survival by 17% (terminal illness 194 dpi treated vs. 166 dpi control). This may be an underestimate of the therapeutic effect because BX912 is highly toxic and reduces lifespan in uninfected mice, so the 17% probably represents some combination of a larger survival benefit plus a toxic effect.
Jean-Philippe Deslys
An unscheduled addition to the program, Deslys conducted the annual business of the NeuroPrion Association. NeuroPrion Association membership is open to all academic labs, scientists, students and governmental organizations for a fee, and to non-profit organizations for free. This gets you access to the private content of the NeuroPrion website, which includes videos of the past conference presentations. He noted that all presentations and data therein remain copyright of the presenters/authors and not of NeuroPrion.
He announced that Prion2015 will be hosted by Glenn Telling in Ft. Collins, Colorado. In addition, Prof. Hidehiro Mizusawa has offered to host Prion2016 in Tokyo, which would be the first time the Prion conference has been in Asia. The members get to vote on whether this will happen, and Deslys is urging people to vote yes.
In the Q&A, Hermann Schatzl asked whether NeuroPrion will start to accept credit cards, and Deslys said yes we’re now accepting PayPal, and I’m all, what on earth was the payment method before?? Gold?
Introduction to the functional genomics section
Gustincich introduced the final block of sessions for the day. There are three lectures on functional genomics approaches to neurodegenerative disease, with the idea of exposing everyone to a new approach that is relatively unfamiliar to the prion audience. He introduced some current concepts in genomics and Thomas Gingeras, who is here to tell us about ENCODE, and Alistair Forrest, who is here to tell us about FANTOM.
Thomas Gingeras
Gingeras began by reminding us that 80% of the genome is transcribed, but most of this is non-coding RNAs. He presented data that most transcripts are expressed very lowly, < .1 FPKM and asked whether/why we care about such lowly-expressed transcripts. His answer: yes, we care, because at the single-cell level, some of these transcripts have no expression in 99% of cells and high expression in 1% of cells. He next questioned our current definition of a gene, because many loci considered to be one gene actually contain multiple (sometimes non-overlapping) transcripts. This has been his message for a couple of years now [Djebali 2012]. He went on to show the strong correlation in expression level between homologous regions of the mouse and human genomes, even in intergenic regions, which is not explained by the degree of promoter sequence similarity.
He then spoke about RNAs in exosomes and microvesicles. He cited Graca Raposo’s and Andy Hill’s work on prions in exosomes [Fevrier 2004, Coleman 2012, Bellingham 2012] to establish the relevance of this work to prions.
Alistair Forrest
Forrest uses Cap Analysis Gene Expression (CAGE) [Shiraki 2003, Kodzius 2006], which is a technique for quantifying promoter and enhancer usage in transcripts. FANTOM has now released an atlas of which promoters and enhancers are being used in which primary cell types and cell lines (though after fiddling with it a couple minutes I couldn’t figure out how to use it). They also released ZENBU, an integrated genome browser and expression profiler.
He said the relevance to disease lies in looking at disease gene expression in relevant cell types, citing [Andersson 2014]. He showed a variety of examples such as HTT, DMPK, and BRCA1, none of which are particularly enriched for higher expression in the affected tissues (respectively CNS, muscle and breast) than in any other tissue. Of genes involved in Parkinson’s disease, some are enriched in the CNS, while others (LRRK2) are not particularly enriched in the CNS. No clear pattern emerged from the examples shown. Noting that PRNP is expressed in many tissues, he suggested that prion disease must either have unreported peripheral phenotypes, or neurons must have factors that confer selective vulnerability.
In Q&A, Simon Mead observed that PRNP is most highly expressed in the CNS and the immune system, and at low levels in other tissues. He asked whether it is common for a protein to have two tissues where it is highly expressed. Forrest responded that this certainly argues prion protein must be important in both of those tissues.
Stefano Gustincich
Gustincich spoke about the functional genomics of Parkinson’s disease. He began by observing that > 70% of protein-coding genes also have an antisense transcript. These sense/antisense pairs may be head-to-head overlapping (on the 5′ ends), tail-to-tail overlapping (on the 3′ ends) or fully overlapping. These pairs are the reason that when we knock out a gene, we are often knocking out an antisense transcript too. Similarly, human diseases where a deletion removes a gene often remove the antisense transcript too.
He listed 10 genes/loci implicated in familial Parkinson’s disease. One of them is UCHL1. His group found, originally in mice, that the homologue Uch-l1 has an antisense transcript which overlaps head-to-head with only 74bp of overlap. They found that the antisense transcript, dubbed AS Uch-l1 is expressed in substantia nigra neurons, and usually localizes in the nucleus. They figured out that the antisense transcript positively regulates the expression of the protein-coding transcript, and that this regulatory activity absolutely requires an inverted SINEB2 element in the antisense transcript, which is a 172bp region which does not overlap with UCLH1. Further, it was known that rapamycin increases UCLH1 and they showed that the antisense transcript is required for this effect, and ΔSINEB2 antisense is actually dominant negative.
The effect is mediated through CAP-dependent translation. When cells encounter stress, they translate proteins more selectively in order to conserve ATP (this is part of the IRES pathway). By inhibiting mTORC1, rapamycin mimics this stress condition, reducing global translation rates but selectively increasing translation of certain proteins including UCLH1 via a CAP complex, and this mechanism requires the antisense transcript.
All this work is published: [Carrieri 2012]. This seems to have been one of the first really compelling stories for a specific function of a lncRNA, and they showed that many other lncRNAs have a similar structure as AS Ucl-h1 does. They call these lncRNAs “SINEUPs” since they contain a SINE2B repeat that UP-regulates translation.
His lab is now working on understanding the structural basis for antisense regulation of transcripts by SINEUPs and on developing RNA therapeutics based on SINEUPs.
Returning to the FANTOM5 database which Dr. Forrest presented, Gustincich showed that their data support antisense transcripts for several genes involved in Parkinson’s, Alzheimer’s and other neurodegenerative diseases. Finally, he showed that in human hCAGE in FANTOM5 there appears to be an antisense PRNP transcript. This is indicated by the trickle of purple (reverse direction) start sites in the hCAGE track, highlighted in the red rectangle below.
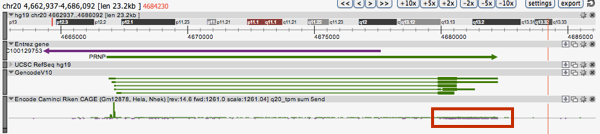
This link should reproduce the above search.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.