The conformation holds the information
As a first-year biology PhD student, I am taking a class called “Analysis of the Biological Literature”, otherwise known as “how to read papers”. One of this week’s papers is the classic prion paper “Conformational variations in an infectious protein determine prion strain differences” by Motamasa Tanaka and Jonathan Weissman [Tanaka 2004]. This landmark work demonstrated that, at least for one yeast prion, strain information is encoded in the physical conformation of the prions. This post is intended to provide a historical context and a full unpacking of the findings of this paper.
Historical context: the state of the prion in 2004
A brief history of the prion hypothesis
When Stanley Prusiner first coined the term prion in 1982, many biologists weren’t happy. He invented this term, a contraction of “proteinaceous infectious particle”, to describe the unique nature of the pathogens that cause a class of diseases such as scrapie, kuru and Creutzfeldt-Jakob disease. The pathogenic agent contained, said Prusiner, “a protein that is required for infectivity” [Prusiner 1982].
In 1982, Prusiner’s principal lines of evidence for the uniquely proteinaceous nature of the pathogen all revolved around what it took to destroy the its infectivity. The infectivity of scrapie, a model prion disease that Prusiner studied in hamsters, could be destroyed by a variety of chemical and enzymatic treatments known to destroy proteins, but was unaffected by a wide range of chemical and enzymatic treatments (and UV light) known to destroy nucleic acids.
Despite initial skepticism, over the next succeeding years, Prusiner and others accumulated a dizzying number of additional lines of evidence which collectively made the existence of prions indisputable. When purified, the pathogen looked like an amyloid made of a polymerized protein [Prusiner 1983]. This protein was dubbed “prion protein” (PrP). Determination of part of its amino acid sequence by Edman degradation [Prusiner 1984] allowed cloning of the gene that encoded it (now called PRNP in humans) [Oesch 1985]. PrP in healthy, uninfected hamsters could be readily degraded by proteinase K but PrP from prion-infected hamsters was partially protease-resistant, suggesting that a post-translational change in this protein might underlie prion pathogenesis [Oesch 1985]. This change appeared to be strictly conformational: the α-helices of healthy “cellular” PrP (PrPC) converted to β-sheets in the “scrapie” form (PrPSc) [Pan 1993]. Mouse lines in which prion disease lasted for different lengths of time proved to have different PrP amino acid sequences [Carlson 1986, Westaway 1987], and PrP knockout mice could not be infected with prions at all [Bueler 1993]. The conformational change in PrP could be reproduced in a cell-free reaction [Kocisko 1994], meaning that the conformational change wasn’t merely secondary to a viral infection or something else. Families with Mendelian forms of prion disease proved to have missense mutations in PRNP that segregated with the disease [Hsiao 1989, Hsiao 1991, Medori 1992] - this existence of a genetic disease that was also experimentally transmissible was unprecedented.
Yet even as the prion concept gained acceptance, the existence of different strains of prions remained difficult to explain. For years this led some researchers to try to propose a role for a nucleic acid in prion disease [e.g. Weissmann 1991], though no such nucleic acid was ever found. By the mid-1990s, it started to seem very likely that prion strain differences were encoded in different conformations of PrPSc. One hint came from the fact that PrPC could acquire the protease resistance of PrPSc in a cell-free reaction [Kocisko 1994], suggesting that prion replication took place via nucleated polymerization, analogous to “ice nine” in Kurt Vonnegut’s Cat’s Cradle [Lansbury & Caughey 1995]. This mechanism finally offered a means by which one could imagine strain information encoded in conformation being transmitted from protein to protein, from organism to organism.
Indeed, by 2004, there were at least a few separate lines of evidence directly arguing that prion strain differences were encoded in different conformations of PrPSc:
- A segment of ~15 amino acids (residues 82-97) in the N terminus of PrP was accessible to proteinase K digestion in prions of some strains but not others. This strain difference could be maintained faithfully even on cell-free templating of PrPSc onto PrPC, suggesting that only prion conformation could be responsible [Bessen 1995].
- Strain differences in protease accessibility and neuropathological profile could also arise spontaneously from different PRNP missense mutations in genetic prion disease, and yet these strain difference were maintained upon transmission of prions to mice [Telling 1996].
- The 3F4 antibody binds to an epitope that is accessible in PrPC and in denatured PrP but buried in all known strains of PrPSc. The amount of guanidine hydrochloride (GdnHCl) required to expose the 3F4 epitope in different strains of PrPSc differs, implying that the resistance to denaturation encoded in the conformation of PrPSc in the different strains must differ [Safar 1998].
But the die-hard skeptics still argued that these experiments all simply proved that differences in conformation were associated with different prion strains, and not that differences in conformation were causal of different strain properties, or in other words, that the conformation was the strain. To such skeptics, the final proof of the conformation-is-strain hypothesis would require generating different conformations of prions de novo, in vitro, from purified protein, and then showing that these prions behaved as different strains in vivo. Indeed, to do so might be hailed as not only the final proof that strain is encoded in conformation, but also the final-final-final proof that prions are comprised of protein. Yet as of spring 2004, it had so far proven impossible to generate de novo prions made of PrP at all, let alone to generate multiple different strains. Luckily, by that time it had also turned out that PrP wasn’t the only prion in nature.
Yeast have prions too
The prion hypothesis turned out to offer a compelling resolution to some longstanding conundrums in the field of yeast genetics.
In the 1960s, through suppressor screens against various yeast mutants, a most peculiar suppressor had been identified [see review and historical narrative in Liebman & Derkatch 1999]. This heritable element, whatever it was, could suppress the phenotypes of several different mutations, mostly with mutations in nutrient biosynthesis pathways [Hawthorne & Mortimer 1963]. Its ability to suppress so many different mutant phenotypes led it to be called a “super-suppressor”. The super-suppressor arose spontaneously in about 1 in every 100,000 cells even absent any mutagenesis, and it occasionally disappeared spontaneously too. It was dominant, and was inherited cytoplasmically, yet could never be linked to mitochondrial DNA, nor to viruses, nor to any other known cytoplasmic element.
A popular system for studying the “super-suppressor” was ade1, an S. cerevisiae mutation disabling a gene in the adenine biosynthesis pathway. ade1 yeast couldn’t grow on adenine-deficient media, which meant you could easily select for suppressors of the mutation. Even when they were grown on adenine-containing media, ade1 yeast were bright red because they accumulated P-ribosylaminoimidazole, a precursor in the adenine biosynthesis pathway (commonly called aminoimidazoleribotide or AIR for short). The ability to do selection plus a phenotype visible to the naked eye meant that ade1 lent itself well to research.
Yeast geneticist Brian Cox used yeast with both the ade1 mutation and the “super-suppressor” element to screen for suppressors of the super-suppressor. It was he who dubbed the whole super-suppressor system “Ψ” (Psi) [Cox 1965, full text]. The intact super-suppressor was called Ψ+ and mutants that suppressed the super-suppressor were called Ψ- mutants. Over time the nomenclature morphed into [PSI+] and [psi-], and the meaning changed slightly too (see below).
Cox posited that the super-suppressor-positive cells “contain a self-replicating particle, or a system of reactions sustaining itself by positive feedback”. If that sounds like a description of a prion, it is. But it took 30 more years and all of the foundations that Prusiner and others had laid studying PrP before the true nature of [PSI+] began to reveal itself. Yeast geneticist Reed Wickner, studying the peculiar properties of the cytoplasmically inherited [URE3] element, presented evidence that [URE3] must be a prion conformation of the Ure2 protein [Wickner 1994]. He reasoned that if yeast had prions, then [PSI+] was probably a prion too.
The truth that eventually revealed itself was as follows. Sup35 is a yeast protein which acts as a translation termination factor (also called a eukaryotic translation release factor), meaning it helps to release the ribosome from an mRNA after the ribosome hits a stop codon. In [PSI+] yeast, Sup35 has aggregated into amyloid fibrils, leaving a dearth of functional Sup35 monomers in the cytoplasm and thus allowing ribosomes to read right through stop codons. The ade1 mutation, it turns out, was a nonsense codon in the middle of the ADE1 gene, and read-through of the stop codon when Sup35 is sequestered in an amyloid state restores translation of a functional protein product. The term [psi-] eventually came to simply refer to any yeast which have soluble monomeric Sup35, not necessarily due to a suppressor-of-the-suppressor mutation.
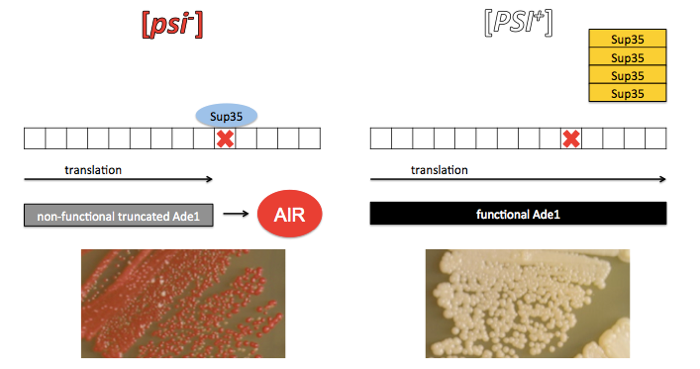
Credit: the photographs of yeast in the above diagram are from Figure 1 of [Bateman & Wickner 2013].
As an aside, you might ask: but isn’t read-through of stop codons bad? Whether prions, including [PSI+], are good or bad for yeast remains controversial. Reed Wickner advocates the view that yeast prions harm their hosts, and that prion domains have evolved divergently to erect prion transmission barriers between different yeast strains [Wickner 2011], while Susan Lindquist champions the notion of yeast prions as being adaptive, and argues that evolution has in fact acted to conserve certain proteins’ ability to form prions [Halfmann & Jarosz 2012].
In either case, the [PSI+] prion proved a facile model system for exploring aspects of the prion hypothesis that had proven fairly intractable for PrP. Years before scientists would succeed at generating synthetic PrP prions, UCSF’s Jonathan Weissman had already in 2000 managed to create [PSI+] prions in vitro from recombinant Sup35 and had successfully infected yeast with them [Sparrer 2000], and had used this system to explore the biochemical basis for “strain barriers” which inhibit transmission of prion states between proteins that are not identical. By 2004, Weissman and his postdoc Motomasa Tanaka were able to generate synthetic prions of different strains, thus finally proving that, for yeast prions at least, the conformation holds the information [Tanaka 2004].
Contributions of this paper
Efficient infection of yeast with synthetic prions
Weissman’s group had previously generated synthetic prions [Sparrer 2000], but had only been able to infect 1-2% of yeast that were exposed, and their protocol didn’t allow them to examine the properties of the synthetic prions they had produced before introducing them into yeast. Their breakthroughs in [Tanaka 2004] relied on a new, higher-efficiency method for introducing synthetic prions into yeast.
The generation of prions was surprisingly simple. They expressed in bacteria a fragment of Sup35 containing the prion-forming domain - this required only the N terminus and the middle of the protein, thus the fragment was called Sup35-NM. They denatured recombinant Sup35-NM protein with guanidine hydrochloride (GdnHCl), and then incubated it in the right buffer conditions and isolated spontaneous amyloid fibrils by centrifugation.
Such a gentle procedure, on its own, tends to create long fibrils with relatively few ends for recruiting new monomers. According to the breakable filament model of prion formation (diagrammed below), PrP and Sup35 prions alike are composed of linear fibrils, where prion propagation requires two steps: elongation, where new monomers are recruited to the ends of fibrils, and fragmentation, where the fibrils are shattered, creating new ends.
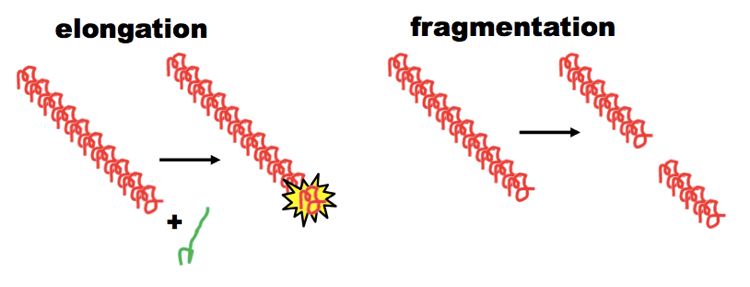
The breakable filament model, diagrammed above, is still considered a hypothesis rather than fact, but it is highly consistent with a lot of observations. For instance, efficient cell-free reactions with PrP usually require sonication or shaking, which are thought to provide the energy for the fragmentation step. In yeast, [PSI+] prion propagation requires having the right amount of the chaperone Hsp104 to cleave the Sup35 fibrils [Chernoff 1995].
Therefore, after creating Sup35-NM fibrils by incubation, Weissman and Tanaka also sheared up their fibrils via sonication, creating many more “ends” for creating new prion infections in yeast.
To introduce the prions into yeast, they first prepared yeast spheroplasts, meaning they removed most of the cell wall, and then they permeablizied the yeast cell membranes with a buffer containing Tris (a detergent to disrupt membrane integrity) and polyethylene glycol (whose purpose remains unclear, at least to me). They guessed that only a fraction of the yeast cells were being successfully permeabilized, and so at the same time as introducing their synthetic Sup35 prions, they also introduced plasmids encoding the URA3 gene. By starting with ura3- and later selecting for yeast that could grow without uracil, they effectively selected for yeast that had been permeabilized and were capable of taking up materials from the surrounding environment - meaning these yeast had at least had an opportunity to become infected with the synthetic Sup35 prions.
This procedure is diagrammed in Fig 1A, and Fig 1B shows what the yeast looked like after plating: negative controls treated with only the URA3 plasmid or with the URA3 plasmid and monomeric denatured Sup35 were all red, while among the cells exposed to URA3 and the synthetic prions, some were white, indicating they had become [PSI+]. Thanks to the URA3 selection procedure, they were able to see a clear dose-response (Fig 1C) - when they added synthetic prions at a Sup35 concentration of 1 μM, they got about 20% of cells to become [PSI+], and when they upped the concentration to 10 μM they got nearly 100% of cells infected. Fig 1C also contains another important control. As noted above, the [PSI+] state can arise spontaneously in yeast at a rate of about 1 in 100,000, but this only occurs when the yeast are already positive for another prion state called [PIN+]. [pin-] yeast are incapable of spontaneously forming [PSI+] prions. In Fig 1C, the authors show that synthetic prion infection was equally efficient in [pin-] and [PIN+] yeast, thus confirming that the [PSI+] transformants they isolated had really been infected with synthetic prions and weren’t just the few cells that had happened to form spontaneous prions.
The authors also showed that their synthetic prions obeyed the laws which Weissman’s group had already worked out for the “species barrier” in yeast prion transmission. It had long been known that PrP prions made of one PrP amino acid sequence had difficulty recruiting PrP monomers of a different amino acid sequence. For instance, it was hard to infect mice with hamster prions, but this barrier could be overcome if the mice expressed hamster PrP transgenes [Scott 1989]. On similar lines, it was thankfully hard to infect humans with cow prions - this is believed to be one reason why only 200 people became infected with variant Creutzfeldt-Jakob disease after exposure to “mad cow” prions in the food supply, even though millions and millions of people in the U.K. were exposed.
The same is true in yeast: Candida albicans Sup35 prions cannot infect Saccharomyces cerevisiae, and vice versa, and one year earlier, Weissman’s group had confirmed that this “species barrier” depended upon the amino acid sequence of the Sup35 [Chien 2003]. In Fig 1E they show positive and negative controls to illustrate their ability to reproduce the species barrier in their lab: prion extracts from S. cerevisiae expressing Sup35 with the S. cerevisiae prion domain amino acid sequence could infect S. cerevisiae expressing the same, but could not infect S. cerevisiae expressing Sup35 with the Candida albicans prion domain. Fig 1D shows that the same is true of the new synthetic prions made of S. cerevisiae Sup35 expressed in bacteria: they can infect S. cerevisiae expressing wild-type Sup35, but cannot infect Candida albicans, and cannot infect S. cerevisiae expressing Sup35 with the Candida albicans prion domain.
Generation of multiple strains in vitro
When Weissman and Tanaka picked colonies of transformed yeast and streaked them out on plates, they found a full spectrum of colors, from bright white to light pink to nearly red (Fig 2A, right panel). By 2004, it was well-known that [PSI+] came in different strains, with “weak” strains recruiting <100% of the monomeric Sup35 into amyloid fibrils, thus allowing some translation-termination to occur. These “weak” strains accumulated intermediate amounts of AIR, and thus appeared pink. Indeed, Fig 2B provides a positive control repeating this experiment with yeast-derived prions of different known strains. The “strong” and “weak” strains in Fig 2B each produce only about 50% successful infections, with some of the streaks being dark red and thus presumably [psi-]. But among the successfully infected colonies, the color is relatively consistent - a bright white in the “strong” strain and a pastel pink in the “weak” strain. Therefore, the fact that the color of colonies transformed with synthetic prions in Fig 2A was so variable indicates that the synthetic prions came in different strains.
Characterization of synthetic prion strains
The fact that the synthetic prions were formed in a cell-free buffer and isolated by centrifugation made it possible to vary the conditions of their formation and then characterize the resulting amyloids before infecting yeast. The researchers found that incubating the denatured Sup35-NM monomers at different temperatures resulted in different biochemical properties. Amyloid aggregates are partially resistant to dissociation by heat and SDS, so when they performed SDS-PAGE on the synthetic Sup35-NM fibrils, the amount of Coomassie-stained monomeric Sup35-NM seen at the expected molecular weight for monomers reflected the degree to which the amyloid had been disrupted. When they formed amyloid fibrils at 4°C, it was easy to get some monomers back simply by heating the fibrils to about 40-60°C in SDS. In contrast, the amyloid fibrils formed at 23°C or 37°C were much more heat-resistant, and you had to heat them to 70°C in order to see any appreciable amount of monomers coming off of the fibrils (Fig 3A).
The resistance of the fibrils to heat and SDS was sort of an indirect measurement of their conformation, analogous to the differential accessibility to proteases or resistance to GdnHCl denaturation seen for different PrP prion strains, as discussed above. Tanaka and Weissman also managed to measure some conformational properties of their amyloid strains directly, using electron paramagnetic resonance (EPR). I don’t understand all the physics of this technique, other than to say it is analogous to NMR, except that it is electrons and not nuclei that are being excited. Like NMR, EPR can give you a readout of how much conformational flexibility different atoms in your molecule have. This technique was used to show that, while the various Sup35-NM amyloids shared broadly similar structures, they had pronounced differences in flexibility at certain residues (Fig 3B-C).
Together, the differences in heat resistance and EPR spectra between the different fibrils argued that the different temperatures had resulted in different prion conformations.
Different conformations result in different strain properties
In Figure 3, the authors had shown that prions formed at different temperatures had different conformations. Next they needed to show that these different conformations constituted different strains - meaning they behaved differently in vivo. They used their protocol again to infect yeast spheroplasts with prions formed at the three temperatures of interest, and they found compelling differences in the results. In Fig 4A, they have streaked out transformants exposed to the 4°C, 23°C, and 37°C synthetic prions, and color is being used as a readout of strain properties. Within the plate for each temperature, there are differences in color reflecting the fact that <100% of clones were infected. But among the infected streaks, notice that the 4°C prions induced many bright white [PSI+] colonies, while the 23°C and 37°C prions resulted in more pink colonies. Some of the 23°C and 37°C colonies are also “sectored”, meaning you can see that part of the streak is pink and part has reverted to red, meaning that the prions had spontaneously disappeared (also referred to as having been “cured”) in some cells. The differential abundance of “pink” versus “white” colonies is quantified in Fig 4B. Together, the pinker color and occasional “curing” of the prions from 23°C and 37°C argued that the prions formed at these higher temperatures were “weak” strains. In addition, Figure S8 shows that it was easier to cure the “weak” strains by over-expressing Hsp104. Though Hsp104 is essential for [PSI+] prion propagation in yeast, its overexpression cleaves the fibrils up so efficiently that it effectively cuts them back into monomers, “curing” the prion state.
{kind=link}
One possible objection was that perhaps the titer of the 4°C prions had been higher, and so the yeast had simply been infected with more prions, and not with a stronger strain of prions. To rule this out, they performed two additional experiments. First, they varied the concentration of prions. They repeated the 4°C infection at 1/10 the original concentration of Sup35 amyloid, and while fewer colonies became [PSI+], the colonies that did do so were still bright white (Fig 4C, left). In contrast, when they repeated the 37°C prion infection at a 10-fold higher concentration, they still got only pink colonies out (Fig 4C, right). Second, they showed that the ratios of pink and white phenotypes generated upon synthetic prion infection were maintained upon serial transmission of prions from yeast to yeast (Fig 4D). The original concentration of synthetic prions could only be expected to influence the phenotype in the first generation of infected yeast, and not in subsequent passages. A note added in proof indicates that they also found that the differences in thermal stability of the prion aggregates were also maintained on serial passage in vivo. Together, these experiments argued that conformation, and not prion titer, determined the strain phenotype.
Conclusions and comparison to other work
This paper provided conclusive evidence that, at least for yeast Sup35 prions, the conformation of a prion encodes its strain properties. For some, this constituted the final evidence for protein-based inheritance, a definitive violation of the central dogma of biology.
Progress in the yeast prion field at that time was rapid, and it is worth noting the distinctions between different breakthroughs. In the same issue of Nature, another group published a paper entitled “Protein-only transmission of three yeast prion strains” [King & Diaz-Avalos 2004]. In that work, the researchers purified prions of different strains from yeast, transmitted the strain properties from those authentic, in vivo-derived yeast prions onto bacterially expressed recombinant Sup35, and then introduced the recombinant Sup35 prions back into yeast, and showed that the strain properties had been maintained. That paper was evidence for (as the title says) the protein-only transmission of strain properties, in contrast to the protein-only generation of distinct strains described by [Tanaka 2004].
Epilogue
This paper resolved a longstanding question in the prion field, by conclusively demonstrating that differences in protein conformation alone were sufficient to generate different strain properties in vivo in yeast. It also raised just as many questions as it answered, including most prominently: what exactly were the conformational differences between the weak and strong strains? Tanaka and Weissman were back two years later with a partial answer to this question [Tanaka 2006]. They presented evidence that, surprisingly, the “weak” strains were actually too stable for their own good. Weak strains were really good at the “elongation” step diagrammed above, but were bad at “fragmentation”. Because they were hard to break, they produced fewer new “ends” to recruit additional Sup35 monomers. Strong strains elongated slowly but were more brittle, and their increased propensity to fragmentation made them propagate more efficiently on the net.
Surprisingly, though, whether conformation is the sole determinant of PrP prion strain has remained controversial. A few months after Tanaka’s paper was published, the Prusiner lab published its first success at generating synthetic PrP prions [Legname 2004], and several years later, the Prusiner lab validated its own results by performing experiments closely analogous to those of [Tanaka 2004]: they generated PrP amyloids under different urea, pH and temperature conditions and then showed that these synthetic prions gave rise to different strain properties when injected into mice [Colby 2009]. Still, many prion scientists objected to Prusiner’s interpretation that prion infectivity and strain properties are contained solely in protein, in part because Prusiner’s synthetic prions, which were prepared from nothing but protein, had very, very low amounts of infectivity. A prion-infected mouse brain, at the time of terminal illness, contains about 109 median infectious units (ID50s), where one ID50 represents the quantity of prions required to produce a lethal prion infection in half of mice exposed. The synthetic prion preparations reported in [Legname 2004] contained roughly on the order of 1 ID50, meaning they were many logs less infectious than authentic, in vivo-derived prions. When other prion scientists eventually created protocols capable of generating tens of thousands of median infectious units of PrP prions, they did so by sonicating their purified PrP together with polyanion and/or phospholipid co-factors [Deleault 2007, Wang 2010]. At least in vitro, such co-factors appear to be essential for creating high titers of prions, as well as for encoding strain properties [Deleault 2012a, Deleault 2012b], suggesting that different PrP prion strains may differ not only in their conformation but in what other co-factor molecules they are complexed with. A final resolution to this controversy continues to prove elusive, and may await the determination of a high-resolution structure of infectious PrPSc.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.