Genetics 27: 'Epigenetics Part II'
These are my notes from lecture 27 of Harvard’s Genetics 201 course, delivered by Fred Winston on December 5, 2014.
This is part II of a two-lecture series on epigenetics. Last time included a general introduction to epigenetics, histone modifications and DNA methylation. This lecture covers imprinting and X inactivation.
The beginning of this lecture finished up last time’s the discussion of DNA methylation - I have added those notes to the notes from last lecture.
Introduction
Imprinting and X-inactivation are both:
- important phenonmena in human biology
- longstanding research areas
- currently popular research areas
- still mysterious in many ways
Some reviews on this subject include [Wilkins & Haig 2003], [Heard 2005], [Zakharova 2009], [Barlow 2011], and [Erwin & Lee 2008].
Imprinting
Imprinting is a phenomenon in mammals where an autosomal gene is inherited in a silent state from one parent and in an active state from the other parent.
In mice, 144 genes are known to be imprinted. In humans, about 100 imprinted genes have been documented. This number can be debated in part because in some cases the “silenced” copy of the gene is not completely silenced, so it depends on your threshold to define a gene as being imprinted. Imprinted genes are involved in a number of human diseases.
The first demonstration of imprinting came from nuclear transfer experiments. In mice zygotes, there exists a moment in time after the sperm enters the egg but before nuclear fusion, where there is a separate male pronucleus and female pronucleus. At this stage, the pronucleus can be transplanted. Transplanting one male and one female nucleus is viable, but transplanting two female or two male pronuclei leads to inviability.
![]()
An additional line of evidence came from translocations, as diagrammed below. Mice have 19 autosomal chromosome pairs plus X and Y, and unlike human chromosomes, all mouse chromosomes are acrocentric, meaning they have the centromere all the way at one end. Thus, chromosomes sometimes fuse near the centromere without loss of genetic information, creating a metacentric chromosome, also known as a Robertsonian fusion (1). A common example is the fusion of 11 and 13. Consider mice heterozygous for the fusion, such that they have one copy of chr11/13, and one copy each of chr11 and chr13 (2). While the chromosomes still can segregate propertly, this fusion scenario results in an increased rate of nondisjunction (3), creating aneuploid gametes. With this setup, a classic experiment was done by [Cattanach & Kirk 1985] and is reviewed and explained further in [Cattanach 1986, full text here]. In this experiment, you cross two mice which are heterozygous for the fusion (4). Because there are six possible gametes from each parent (see 3), the Punnett square for this cross (5) is huge. Three outcomes are possible. Some offspring will get one copy of each chromosome from each parent (smiley faces) which is not interesting. Others will get trisomy or monosomy of at least one chromosome, which is lethal (red X). Yet other offspring will be balanced (diploid for every chromosome), but have uniparental disomy of one chromosome (UD). To can use recessive markers to distinguish these from the normal offspring. Phenotyping these offspring (6) revealed that for one chromosome, mice with balanced maternal disomy had reduced size, while the mice in with balanced paternal disomy had increased size. This was then done systematically for every chromosome, revealing only ~10 regions, each containing several genes, which were subject to imprinting. Some imprints were male-specific and others female-specific.
![]()
Mechanistically, you can reason that the imprint must be:
- established in the gametes
- maintained during development
- erased and established during germ cell development
The first connection to human disease came from the characterization of Angelman syndrome in 1965 by a British pediatrician. In the U.S. Angelman syndrome is found in ~1 in 20,000 live births. It is caused by mutations in the UBE3A, which encodes a ubiquitin ligase. About 75% of cases are associated with a large 6Mb deletion on chr15 including UBE3A. This in itself was not evidence for imprinting - there are plenty of deletions of this size or smaller which cause severe diseases simply because they include at least one haploinsufficient gene. ~15% of cases have point mutations in UBE3A, which narrows it down to that gene. Analysis of Angelman syndrome pedigrees revealed that the condition manifests in a child only when transmitted from a female carrier, not from a male carrier, as shown in this pedigree chart.
This unusual inheritance pattern provided evidence that imprinting was involved. Only the maternal allele of UBE3A is expressed in neurons, while both alleles are expressed in other cell types. This is due to a long non-coding antisense RNA whose expression is controlled by a cluster of DNA methylation:
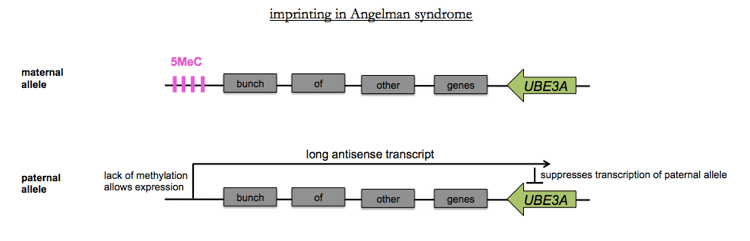
This has led to a new potential therapy for Angelman syndrome, currently in preclinical development: using ASOs to knock down the long antisense RNA, thus allowing expression of the paternal allele [Meng 2014].
Prader-Willi syndrome is associated with another imprinted region nearby, where only the paternal allele is expressed, and not the maternal allele.
We still don’t entirely know why nature evolved imprinting; some theories are reviewed in [Wilkins & Haig 2003].
X-inactivation
Males and females have different numbers of X chromosomes, but express approximately the same level of X-encoded information, indicating there is some mechanism for dosage compensation.
In different organisms this is acheived in different ways:
| organism | male | female | dosage compensation mechanism |
|---|---|---|---|
| flies | XY | XX | elevation of X expression in males |
| worms | XO | XX | repression of X in hermaphrodites |
| mice | XY | XX | random inactivation of 1 of 2 X chromosomes in females |
| humans | XY | XX | random inactivation of 1 of 2 X chromosomes in females |
The fact that flies and worms had opposing mechanisms was originally worked out through genetic analysis of mutants.
In mammals, X-inactivation:
- Is the transcriptional silencing of almost an entire X
- Occurs early in the development of female embryos, at the ~64 cell stage
- Occurs at random, but is subsequently stable in each cell for the rest of the organism’s lifespan
As a result, adult female mammals are mosaics.
X-inactivation was first described by [Barr & Bertram 1949]. They noted a dark staining of a condensed body at the nuclear periphery only in female cells. Later, Mary Lyon in 1961 proposed the from random inactivation of one X chromosome early in development. Xi came to refer to the inactive chromosome and Xa to the active one.
Xi has several unique features:
- undergoes late DNA replication
- has more condensed chromatin (hence the “Barr body” appearance)
- is enriched for repressive histone modifications
- has a distinct pattern of DNA methylation
- most genes are completely silenced
- is coated in a 17kb lincRNA called Xist
X inactivation requires several steps:
- Establishment of the Xi requires counting, choice and spreading. Counting obeys an “n-1 rule” ensuring that the number of inactive Xs is the total number of Xs minus 1. This is trivially obvious in XY and XX genotypes, but females with three X chromosomes (XXX) turn out to have 2 inactive Xs. Once an inactivation signal originates on one chromosome, silencing spreads in cis, mediated by Xist RNA.
- Maintenance of the Xi requires that its silenced-ness is transmitted upon every cell division.
- Erasure of the silencing is performed in gametes so that either X can be active upon transmission to a zygote.
Xist is expressed only in females, does not encode a protein, coats the Xi and is required for establishment but not maintenance of Xi.
Heterozygous deletion of Xist in ES cells results in a situation where the ΔXist X chromosome never inactivates, while the wild-type chromosome, designated X+, sometimes inactivates. Thus, Xist is required for inactivation but not for choice. It turns out that there is also a 40kb antisense RNA whose genomic sequene is a superset of Xist’s, which is called Tsix and which negatively regulates Xist. There are many other non-coding RNAs which are also involved in ensuring that only one X gets silenced.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.