Genetics 26: 'Epigenetics Part I'
These are my notes from lecture 26 of Harvard’s Genetics 201 course, delivered by Fred Winston on December 3, 2014.
This is part of a two-lecture series on epigenetics. Today will include a general introduction to epigenetics, histone modifications and DNA methylation. The next lecture will cover imprinting and X inactivation.
Introduction
Until now, we’ve been told that a genotype is the state of a DNA sequence. In other words, a wild-type phenotype is associated with a wild-type sequence, while a Mendelian mutant phenotype is associated with a mutant sequence, and a complex mutant phenotype is associated with many sequence changes. However, sometimes inheritance of a phenotype is determined by factors other than DNA sequence. Such factors are referred to as epigenetic. We now know that units of inheritance also include (but are not limited to):
- DNA sequence
- DNA modifications
- Proteins and RNAs bound to DNA
Where items 1-3 comprise chromatin. Item 3 includes:
- histones
- tautologically, “non-histone proteins” (which include transcription factors, repressors, etc)
- ncRNAs such as XIST
These factors can in turn be modified by:
- environmental factors
- the chromosomal position of a gene
- the nuclear position of a gene
- interactions between homologous genes
Units of inheritance are not limited to chromatin either - consider also prions in yeast and dsRNA propagated by RNA-dependent RNA polymerase in C. elegans, though this series of two lectures will only cover chromatin.
It is currently controversial how much is included in the term “epigenetics”. For example, some histone modifications exhibit clear transgenerational inheritance and thus definitely qualify as epigenetic, while other histone modifications seem to be established de novo in each individual, so some investigators argue these should not be categorized as epigenetic.
Two useful references are [Brookes & Shi 2014, Ho 2014].
Examples
Gene silencing in yeast
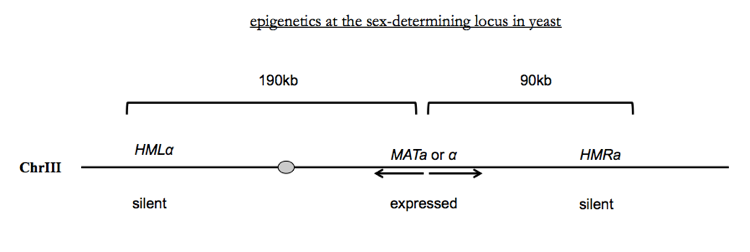
Recall that yeast have two mating types dubbed a and α, and the gene complex determining mating type covers ~317kb on yeast chromosome III. The MATa/MATα locus includes genes transcribed in opposite directions from a common starting point. 190kb to the left is a locus HMLα and 90kb to the right is a locus called HMRa. These two loci encode, respectively, all of the same machinery as MATa and MATα do, but in the normal state of yeast, the MAT locus is expressed while the other two loci are silenced. This silencing is accomplished by repressive histone modifications and by a class of non-histone proteins called Sir proteins. Sir proteins have orthologs in mammals called Sirtuins which regulate metabolism, mitochondrial function and so on.
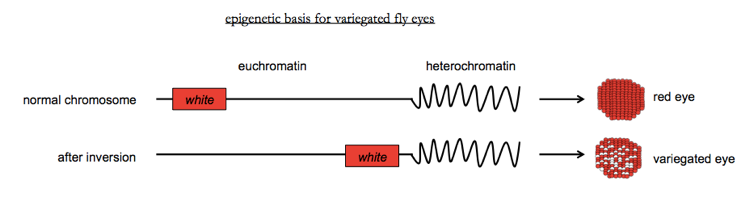
Variegation in flies
Wild-type Drosophila have red eyes; the gene which encodes the red pigment is confusingly called white, after its knockout phenotype. A chromosomal inversion which moves the wild-type white gene closer to heterochromtain results in variable silencing of the gene in some cells but not others, causing a phenotype in which some but not all cells of the eye lose their red color. This formed the basis of some enhancer screens in Drosophila and helped to reveal some of the first histone methytransferases which act on H3K9.
Reprogramming of somatic cells
Somatic cells can be reprogrammed to induced pluripotent stem cells (iPSC) by transient expression of SOX2, OCT4, KLF4 and MYC. This can be accomplished by transfection with only the mRNAs, or expression of a non-integrating virus (e.g. Sendai) or an integrating virus (lentivirus). Typically only 5% of cells exposed to the reprogramming factors will successfully revert to pluripotency.
Phage λ-infected E. coli
The lysogeny vs. lytic replication decision is controlled epigenetically.
Drosophila sex determination
Cellular sex determination in flies is determined through chromatin.
Honeybee queen/worker determination
Compare and contrast the queen and worker bees:
| bee | food | lifespan | fertility |
|---|---|---|---|
| workers | pollen + nectar | weeks | sterile |
| queen | royal jelly | 1-3 years | up to 2000 eggs/day |
A breakthrough paper showed that RNAi against DNMT3 - a DNA methyltranferase - caused most larvae to develop as queens [Kucharski 2008].
Amusingly, Fred notes that royal jelly is available for human consumption on Amazon.
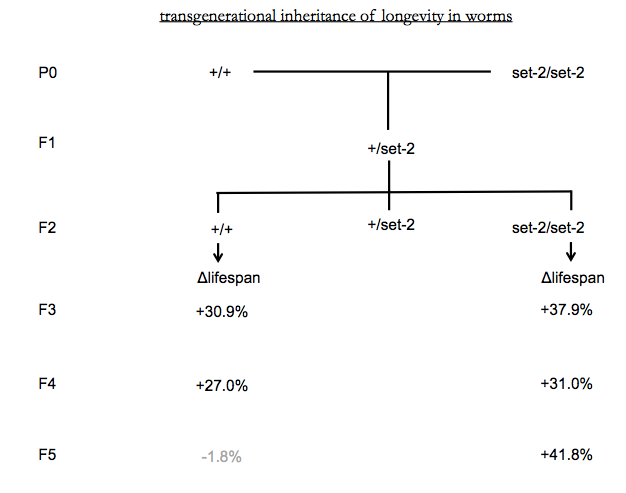
Lifespan in C. elegans
Mutations in set-2 in C. elegans decrease H3K4me3 marks and increase longevity by ~30%. This phenotype is then inherited transgenerationally even after crossing to remove the originally causal genetic mutation [Greer 2011]. The increase in lifespan remains significant through the F4 generation of wild-type descendants (see below diagram) and finally disappears by F5.
Histone modifications
Recall from Timur Yusufzai’s molecular biology lecture 15 that histones are small proteins, ~100 amino acids. They are subject to a wide variety of post-translational modifications including but not limited to: acetylation, methylation, ubiquitylation, sumoylation and phosphorylation. Usually these modifications occur in the N-terminal tail, which is roughly residues 1-25. Occasionally you’ll hear of a modification to the C-terminal core of the histones, usually at K56 or K79. The notation is as follows: H3K4me3 means histone 3 lysine 4 trimethylation.
Some of the most important modifications are on histone 3:
| modification | association |
|---|---|
| H3K4me3 | active transcription |
| H3K9me3 | repressed transcription |
| H3K9Ac | active transcription |
| H3K27me3 | repressed transcription |
The ENCODE project is an effort to systematically map histone methylation (among other things) in humans [ENCODE 2012]. There are comparable efforts in other organisms, such as mod-ENCODE in Drosophila and C. elegans. These have been largely descriptive efforts. For instance, in Drosophila they’ve now mapped 18 histone modifications and 9 combinatorial patterns, yet we still do not know how the different domains form, what their roles are, how they differ between cell types, or what the cause-effect relationship is between these and other findings. Dissecting the functional relationships will require interventions rather than just observations - for instance, experiments using RNAi.
DNA methylation
DNA methylation is the chemical modification of cytosine to 5-methylcytosine (variably abbreviated 5mc, 5mC or 5MeC).
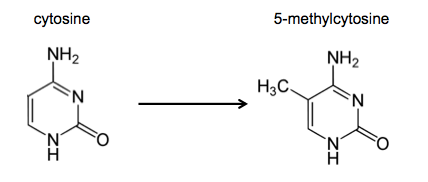
This occurs (almost?) exclusively in a CpG dinucleotide context. In mammals, 70-80% of cytosines CpG dinucleotides are 5-methylcytosines.
Some organisms have DNA methylation, while others do not:
| organism | DNA methylation |
|---|---|
| E. coli | yes |
| yeast | no |
| fungi | yes |
| worms | no |
| flies | a little |
| mammals | yes |
| zebrafish | yes |
| plants | yes |
DNA methylation regulates gene expression by several mechanisms:
- It affects the binding of transcription factors
- Some proteins specifically bind 5mc
- It can “act with histone modifications”
Several enzymes control DNA methylation. DNMT3a and DNMT3b recognize CpG dinucleotides and methylate the C on both strands.
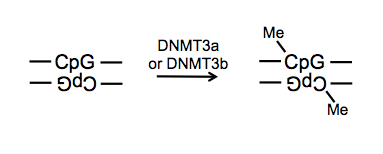
In mice, DNMT3a and DNMT3b:
- are required for new DNA methylation
- are each essential for normal development
- act over different genomic regions than each other
- interact primarily with the tails of H3 that lack K4 methylation
In humans, DNMT3b mutations cause a recessive disease called ICF syndrome [OMIM #242860].
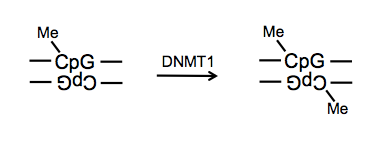
After DNA replication occurs, dimethylated CpG sites (as created by DNMT3a/b) will end up hemimethylated. A different enzyme, DNMT1, is responsible for then returning these to a dimethylated state. DNMT1 knockout is embryonic lethal.
Assays to study DNA methylation
- Restriction enzymes HpaII and MspI both cut CCGG sites, but MspI cuts regardless of methylation state, while HpaII cuts C-C-G-G but not C-5mC-G-G.
- ChIP and ChIP-seq can be performed using antibodies specific for 5mC.
- Sodium bisulfite deaminates only unmethylated Cs, converting them to Us, in denatured DNA. You can therefore treat genomic DNA with sodium bisulfite, then PCR-amplify a sequencing library, and compare the sequencing reads to those from the same genome when sequenced without bisulfite. Bases that are C in either library represent methylated Cs, while C→T SNPs that are unique to the bisulfite-treated library are unmethylated Cs. This procedure and its pros and cons are explained in more detail in my notes from the UAB Sequencing Short Course.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.