Genetics in medicine 02: Mitochondrial disorders
These are my notes for week 2 of Harvard’s Genetics 228: Genetics in Medicine from Bench to Bedside course. I added the class late so this is just a quick review of the paper discussed in class.
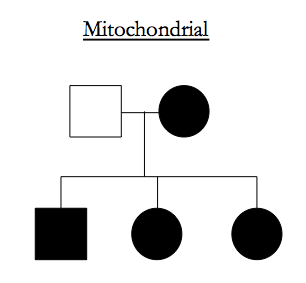
It is important to distinguish mitochondrial disorders from a mitochondrial inheritance pattern. As introduced here a mitochondrial inheritance pattern is one in which affected mothers transmit to all children, while affected fathers do not transmit:
Diseases exhibit this inheritance pattern† if they are caused by variants located in the 16.6kb of human mitochondrial DNA. However, there are also about 1,600 nuclear genes with mitochondrial responsibilities [Lieber 2013]. Mutations in these genes lead to autosomally inherited defects in mitochondrial metabolism.
†An important caveat to the overly simplistic above diagram is that many people are at least somewhat heteroplasmic - they have mitochondria with a few different mitochondrial chromosomes floating around. Therefore the phenotype can be modified considerably by the percentage of mutant mitochondria that a child receives from an affected mother.
The paper for this week [Tucker 2011] reports the discovery of a new mitochondrial disease caused by mutations in MTFMT on chromosome 15. Each patient presented with Leigh syndrome [OMIM #256000], which is the blanket term for a variable collection of neurological signs secondary to defects in oxidative phosphorylation, otherwise known as the electron transport chain. The authors performed targeted capture and seqeuncing of what they called the “MitoExome” — 1,034 nuclear-encoded genes believed to be involved with mitochondria. They did the usual functional filtering for predicted deleterious functional classes and for variants with an allele frequency of <0.5% in “public databases” (presumably 1000 Genomes and ESP at that time). After these filters, MTFMT was the only gene with a potentially recessive inheritance pattern. Each patient was compound heterozygous, with a splice enhancer site mutation at the -20 position of exon 4, and either a nonsense (R128X) or missense (S125L) variant in trans.
It is pretty incredible that the authors were able to identify this gene as causal in these cases. In the Mendelian variant discovery efforts that I was involved with in the MacArthur Lab, even “extended” splice site mutations (3 to 8 bases from the splice site) were nearly always benign false positives, and the more distal splice enhancer regions rarely even had enough coverage to get good genotype calls. This mutation would be very likely to be missed in whole-exome sequencing. Therefore, the ability to narrow down the target to about 1,000 genes thanks to strong biological candidacy of mitochondrial defects was probably an important part of what enabled these cases to be solved.
MTFMT’s protein product has an interesting biological role that I didn’t know anything about until reading this paper. It covalently modifies the methionine in mitochondrial Met-tRNAMet to make it N-formylmethionine. In mitochondria, this modified amino acid is selectively used for translation initiation, whereas regular methionine is used for translation elongation. To convince themselves that these mutations were causal, the authors demonstrated exon skipping in MTFMT with qPCR (Fig 2C), and showed that the patients’ cells had a defect of methionine formylation (Fig 3) resulting in reduced translation of some mitochondrial genes, which they were able to rescue upon transformation with lentivirus encoding wild-type MTFMT (Fig 2D).

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.