Backgrounder on SOD1
This semester I’m doing a research rotation in Lee Rubin’s lab where they have been modeling SOD1 ALS mutations in iPS-derived motor neurons [Yang 2013], and I’ve also decided to do my final project for my protein folding class on the prion-like aspects of SOD1 misfolding. This post is my first pass at a backgrounder to get familiar with the gene, the protein, and just a little bit of the literature.
Background on the gene and the protein
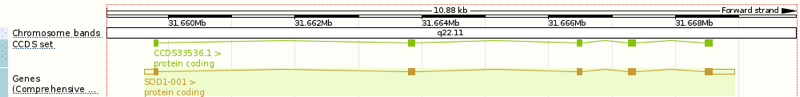
Above: the SOD1 gene, ENSG00000142168 in the Ensembl viewer.
The SOD1 gene (above) spans about 10kb of genomic sequence on chromosome 21, with five protein-coding exons encoding a 153-residue protein:
>ENSG00000142168:ENST00000270142 peptide: ENSP00000270142 pep:KNOWN_protein_coding
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS
AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV
HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Note that the translated sequence above is 154 residues, but like many cytosolic proteins [Giglione 2004], SOD1 undergoes N-terminal methionine excision, thus yielding a 153-residue protein. The residue numbering in common use in publications about SOD1 is based on 153-residue counting and thus appears as shifted from this reference sequence by -1: for instance, people refer to D90A [Al-Chalabi 1998] and G93A [Yang 2013], which correspond to residues 91 and 94, respectively, in the above sequence or in any output generated programmatically by genomic tools. In ExAC, the SOD1 gene (canonical transcript ENST00000270142) has three predicted loss-of-function variants, all singletons. The protein is localized to the cytosol and the mitochondrial intermembrane space [Kawamata & Manfredi 2010]. SOD1 was known as an enzyme responsible for inactivating superoxide radicals before it was ever linked to ALS, and its crystal structure was solved in 1982 [Tainer 1982, PDB# 2SOD]. It both functions in vivo, and crystallizes, as a dimer:
fetch 2sod
bg_color white
hide everything
show cartoon, chain O or chain Y
color orange, chain O
color yellow, chain Y
show spheres, inorganic and (chain O or chain Y)
color 0xC77F33, element Cu
color 0x7C7FAF, element Zn
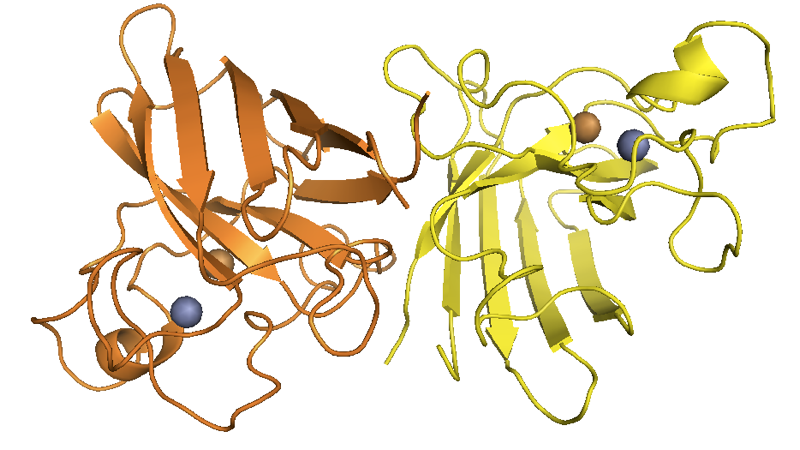
Above: SOD1 dimer structure as solved by [Tainer 1982], with the official PyMOL element colors for Zn and Cu
Many later versions of the structure have been solved as well (e.g. [PDB# 1SOS, Parge 1992]).
I blogged about ALS last semester in these notes from neurodegeneration seminar and an important point is that ALS is the name for a constellation of clincial signs, a syndrome. The name ALS refers to what are probably several different diseases at the molecular level. Most cases are sporadic; of the cases that are familial, there are a variety of genes, often with distinct pathology.
The SOD1 gene was linked to ALS around the same time by two different groups, at MGH and Northwestern [Rosen 1993, Deng 1993], each of which found different mutations in different families, and estimated that 10-15% of familial ALS cases were attributable to this gene†. The first paper remained fairly agnostic as to the mechanism by which these mutations caused disease, offering that “one possibility… is that the activity of SOD1 is reduced, leading to an accumulation of the toxic superoxide radical. Alternatively, the activity of SOD1 might be increased, leading to excessive levels of hydrogen peroxide and the highly toxic hydroxyl radical…” [Rosen 1993]. The other paper concluded more strongly that loss-of-function was responsible - they measured the enzymatic activity of the SOD1 mutants and found it to be on average to be less than half that of wild-type SOD1, and examined the position of the mutations in the reported structure of SOD1 and found them to be disruptive [Deng 1993].
†There is now some evidence that wild-type SOD1 may also be involved in subset of sporadic ALS cases [Bosco 2010, Forsberg 2010], though this remains controversial [Kerman 2010].
The loss-of-function hypothesis was challenged the following year, however, by David Borchelt, who was then working in Don Cleveland’s lab. They found that the G37R mutant retained nearly full enzymatic activity [Borchelt 1994]. To explore the disease mechanism further, they created a whole series of transgenic mouse lines [Wong 1995]. The higher the level of overexpression of G37R SOD1, the earlier the onset of disease in mice; and this was on a wild-type background, so endogenous mouse Sod1 was present. Meanwhile, overexpression of wild-type SOD1 did not cause any pathology. Collectively, these results indicated that the disease might be caused not by loss-of-function, but by “an aberrant property of this SOD1 mutant”, or in other words, gain-of-function.
Further animal data would soon validate this conclusion. Sod1 knockout mice are viable and show no obvious ALS-like phenotype [Reaume 1996], and indeed, the disease course of mice expressing mutant SOD1 is unaltered whether the endogenous Sod1 alleles are present or knocked out [Bruijn 1998]. There is, however, still some controversy as to whether loss of SOD1 function might contribute somewhat to the disease in SOD1 ALS patients [Saccon 2013].
Fast-forwarding 15+ years, mutant SOD1 is now widely considered - depending on your semantic opinion - a “prion” [Prusiner 2012], “prion-like” [Polymenidou & Cleveland 2011], or a “prionoid” [Aguzzi & Rajendran 2009], and is said to cause a “protein misfolding disease” [Nordlund & Oliveberg 2008], or a “protein aggregation disease” [Ray 2005]. As is the case in most (all?) neurodegenerative diseases, it remains controversial whether the toxic species is the aggregate or an elusive oligomer [Polymenidou & Cleveland 2011], but in either case, the death of neurons is ascribed to the progressive, nucleated misfolding of SOD1 protein.
Reading list
My mentor in the Rubin lab, Chen Benkler, gave me a reading list of six of her favorite papers on SOD1, including one review [Polymenidou & Cleveland 2011] and five original research papers focusing primarily on the prion-like spreading of misfolded SOD1 [Matsumoto 2005, Karch 2009, Prudencio 2009, Munch 2011, Basso 2013]. Here’s my read of each of these papers.
The Seeds of Neurodegeneration: Prion-like Spreading in ALS [Polymenidou & Cleveland 2011]
Table 1 compares different neurodegenerative disease-associated proteins in terms of the weight of evidence in favor of a “prion-like” mechanism in different experimental paradigms. At least as of 2011, SOD1 had been shown to undergo seeded aggregation in vitro and in cell culture, and to transmit from cell to cell in cell culture (see [Prudencio 2009, Munch 2011] reviewed below). This would make it comparable to huntingtin in terms of level of evidence, and less strong than Aβ or tau, both of which have been shown to induce aggregation in vivo (in mice), which in turn are less strong than α-synuclein, aggregates of which can actually cause clinical disease in mice [Mougenot 2012, Luk 2012, Watts 2013]. There has been some progress in this area since this review was written, however: last year it was reported that spinal cord homogenates from SOD1 G93A (but not G37R) mice could induce disease in mice expressing a different transgene (G85R-YFP) [Ayers 2014].
The most interesting thing I learned from this review is that SOD1 toxicity is considered to be non-cell-autonomous. This was demonstrated through a series of experiments from Don Cleveland’s lab in which mice with a floxed G37R SOD1 transgene were crossed to mice expressing Cre recombinase under different cell type-specific promoters, to selectively delete SOD1 in either motor neurons, astrocytes or microglia. I coarsely summarize the results in this table:
| Cre promoter | cell type in which SOD1 is deleted | delay in onset | extension of survival | citation |
|---|---|---|---|---|
| Isl1 | motor neurons | 9% | 22% | [Boillee 2006] |
| CD11b | microglia | none | 34% | [Boillee 2006] |
| GFPA | astrocytes | none | 16% | [Yamanaka 2008] |
These results imply that SOD1 in each of these three cell types contributes to the disease, and that SOD1 in microglia and astrocytes affects disease progression but not onset. The authors conclude that there are two distinct disease phases mediated by these different cell types
an initiating phase of disease caused by mutant SOD1 damage within motor neurons, and a mechanistically divergent later phase encompassing the progression to complete paralysis that is linked to the inflammatory response of microglia and mutant toxicity within these cells
— [Boillee 2006]
The finding that mutant SOD1 toxicity is non-cell-autonomous has been further replicated by transplanting glia expressing mutant (G93A) SOD1 into wild-type rats, where they caused motor neuron death [Papadeas 2011]. What’s more, astrocytes derived from iPS from genetic or sporadic ALS patients kill motor neurons when the two are co-cultured [Haidet-Phillips 2011].
Boillee’s notion of “two distinct disease phases” may sound familiar from the study of prion disease - see this post. Yet the situation in prion disease is starkly different from that in SOD1 ALS: neurons that don’t express PrP are simply invulnerable to prion neurotoxicity, period [Brandner 1996, Mallucci 2003]. And although microglial activation and astrocyte proliferation are prominent features of prion disease, the evidence is mixed as to whether these glial cells are friend or foe [Aguzzi 2013]. For instance, deletion of Mfge8, which astrocytes produce and which appears to be involved in apoptotic body engulfment, slightly accelerates prion disease progression [Kranich 2010], whereas deletion of Nox2, which microglia use to produce free oxygen radicals, slightly slows prion disease progression (as does killing off the microglia themselves) [Sorce 2014]. Meanwhile, deletion of Trem2 reduces microglial activation without appreciably affecting disease course [Zhu 2015].
As a prion scientist, something that bothers me about all of the SOD1 toxicity studies above is that they were done on a wild-type background, i.e. endogenous Sod1 was expressed in all of the cells being studied. If prions composed of mutant SOD1 are capable of templating the misfolding of wild-type SOD1, then it is hard to disentangle the effects of prion propagation (which is non-cell-autonomous) from those of neurotoxicity (which the authors conclude is non-cell-autonomous). This issue apparently bothered prionologist Neil Cashman as well, so he studied cross-seeding between mutant human SOD1, wild-type human SOD1 and wild-type mouse SOD1 in cell culture and in vitro and concluded that the mutants could template wild-type human SOD1 but not wild-type mouse SOD1 [Grad 2011]. If so, that’s not so different from the situation with PrP prions, where at least human PrP mutants appear to template the wild-type human protein (see this post), yet a substantial transmission barrier exists between species [Collinge & Clarke 2007]. Still, I’d feel more comfortable reaching strong conclusions about cell autonomousness if the in vivo experiments were repeated on a knockout background.
Structural properties and neuronal toxicity of amyotrophic lateral sclerosis–associated Cu/Zn superoxide dismutase 1 aggregates [Matsumoto 2005]
In this study, they expressed YFP or CFP fused to wild-type, G85R or G93A SOD1 (six combinations total) by transient transfection in rat PC12 cells and then did microscopy, biochemistry, FRAP (see here), and enzymatic activity assays on them. The mutants fused to CFP formed aggregates described as “perinuclear”, which were relatively immobile (they don’t move much according to FRAP), while a subset of the mutant fusion protein was still diffuse and mobile (i.e. <100% of the protein was incorporated into aggregates).
When they co-expressed a mutant SOD1 fused to CFP, and then separately expressed YFP, they found that the the YFP diffused throughout the cell including the space occupied by the mutant-CFP aggregates, which was taken to imply that the SOD1 aggregates form a “honeycomb”-like structure that allows soluble proteins to diffuse through. In contrast, when they expressed a mutant huntingtin fragment fused to CFP, it formed aggregates that excluded YFP. Yet when they coexpressed mutant SOD1-CFP fusions and also the chaperone Hsp70 fused to YFP, the Hsp70 was found to co-localize partly (34%) with the CFP-marked aggregates. This was taken to mean that Hsp70 interacts with the aggregates.
They also tried tagging the LMP2 β subunit of the 20S proteasome with YFP, and they found that the YFP in this case showed 72% co-localization with CFP, implying that the proteasome is sequestered by the SOD1 aggregates. To explore the functional consequences of this sequestration, they co-transfected mutant SOD1-CFP with a YFP-ubiquitin reporter protein. They found that the reporter accumulated to ~2.6 to 2.9x higher levels in the cells containing CFP aggregates, suggesting a reduction in proteasome activity, which they figure is due to the sequestration, though if real the effect could also simply be due to the proteasome being busy degrading the mutant SOD1.
Finally, they make the case that aggregates kill cells. The trouble is that not all cells that are transfected with mutant SOD1-CFP actually develop aggregates - only 15-30% do. So the comparison they end up making is that ~15% of untransfected or WT SOD1-CFP transfected cells died after 48h, while 90% of the cells that were transfected with mutant SOD1-CFP and developed aggregates died. Over that 48h time period, yet additional cells that initially had diffuse mutant SOD1-CFP expression develop aggregates, and 70% of those cells then die during the next 48h.
Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS [Karch 2009]
This paper from David Borchelt’s group studies aggregates obtained from three different transgenic mouse lines expressing human SOD1 with either the G93A, G37R, or H46R+H48Q mutations. They extracted spinal cords in non-ionic detergent and plotted the amount of detergent-insoluble SOD1 immunoreactivity (corresponding to aggregates) over time. While the kinetics of disease were somewhat different in the different mouse lines, the overall picture was similar: the amount of aggregated SOD1 increased over time.
Other groups had previously reported that SOD1 aggregates (unlike natively folded SOD1) are cross-linked by intermolecular disulfide bonds [Jonsson 2004, Jonsson 2006, Furukawa 2006]. Karch et al therefore examined cross-linking in the SOD1 aggregates from these mice. This required detergent extraction of spinal cords in the presence of iodoacetamide (IA), which “irreversibly prevents air oxidation and disulfide bond scrambling during sample preparation” - in other words, making sure that the state of disulfide bonds by the time the protein gets to your gel is the same as what it was in vivo. By using 1% SDS they were able to soluibilize the SOD1 aggregates (which had been insoluble in 0.5% Nonidet P-40 non-ionic detergent) to the point that they could run them on a Western blot and assess their multimericity by molecular weight. Predictably, they found that the appearance of dimers and higher-order multimers increased over the course of disease. They also found that some of the mutant protein migrates on a gel in a manner consistent with a reduced (i.e. not disulfide-bonded) SOD1 monomer, suggesting that perhaps some of the mutant protein had either lost its native (intramolecular) disulfide bond, or perhaps never acquired it in the first place.
This is an interesting contrast to PrP prions. The hypothesis that PrPSc might contain intermolecular disulfide bonds was explored some time ago [Welker 2001] but no one ever managed to produce convincing evidence for it. At a talk that Byron Caughey gave recently, an audience member asked about the possibility of intermolecular disulfides in PrPSc and Byron replied that he had looked into this for a while many years ago but found no evidence for it and so gave it up. Indeed, the Prusiner lab’s efforts 22 years ago failed to find covalent modifications of any type associated with prions, suggesting that PrPSc instead differs from PrPC solely by conformation [Pan 1993, Stahl 1993]. There have been occasional challenges to this view [e.g. Canello 2008] but to my knowledge, the prevaling view among prion scientists continues to be that PrPSc and PrPC differ only in conformation.
Variation in aggregation propensities among ALS-associated variants of SOD1: correlation to human disease [Prudencio 2009]
This is an interesting combination of biochemistry and epidemiology, looking at 21 different SOD1 mutations. They estimate “aggregation propensity” by transiently transfecting HEK293FT cells with the different mutant cDNAs, centrifuging the cell lysates, and then dividing the Western blot band intensity of the pellet (insoluble fraction) by the supernatant (soluble fraction). They then compare this “aggregation propensity” to a bunch of different clinical variables, to see how the aggregation propensity affects the disease. None of the variables of interest were found to correlate with the aggregation propensity. Average age of onset (which varies from about 40 to about 55 between different mutations) was completely uncorrelated with aggregation propensity (p = .92) and disease duration showed only an extremely weak trend (p = .14). They also compared the aggregation propensity to a number of different biochemical factors to see if they could determine what drives aggregation, but none of those correlated either.
Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells [Munch 2011]
This study looks at the ability of cultured cells to take up and propagate SOD1 aggregates. They purified recombinant mutant SOD1 from Sf9 cells (details in [Munch 2010]), labeled it with Dylight649 and then mixed it with trifluoroethanol, which apparently causes it to aggregate. Then they dumped the aggregates onto N2a cells. The N2a cells took up the fluorescent aggregates amazingly quickly - fluorescence inside cells was visible within 60 minutes and then increased by a few orders of magnitude, peaking around 15h. They did this originally with the mutant H64R but replicated the results with two other mutants, A4V and L144F.
They next asked how the aggregates get into cells. They started with FM 4-64FX, which is apparently a dye that stains endocytic vesicles. 5 minutes after exposure to cells, the aggregates appeared punctate and co-localized with that marker, but by the 30 minute mark they appeared to have diffused throughout the cytosol. Thus, apparently the aggregates get endocytosed but then somehow break out of their vesicles. By what pathway, exactly, were the aggregates entering cells? They did a battery of tests to rule out different possibilities. The aggregates didn’t co-localize with transferrin, and their uptake was not inhibited by chlorpromazine, nor by either of two dominant-negative proteins (eps15 and AP180), so the authors ruled out clathrin-mediated endocytosis. A dynamin inhibitor and a dominant negative K44A dynamin mutant did not affect the process, either, thus ruling out dynamin-mediated transport. Caveolin-1 knockout or genistein (a caveolin-1 inhibitor) didn’t affect it either, ruling out caveolin-dependent endocytosis. Yet depleting cellular ATP with sodium azide and 2-deoxyglucose did reduce the uptake of aggregates, indicating there was an active (ATP-driven) process at work.
Finally, they found that 5-N-ethyl-N-isopropyl-amiloride (EIPA), wortmannin, and another compound called IPA3 - all small molecule inhibitors of kinases involved in macropinocytosis - all reduced aggregate uptake. Macropinocytosis is introduced here and has also been implicated in cellular uptake of tau aggregates [Holmes 2013]. Methyl-β-cyclodextrin (MβCD), which disrupts lipid rafts, also reduced the uptake, implicating macropinocytosis at lipid rafts in particular.
Then they asked whether the aggregates could seed new aggregate formation inside cells. They stably expressed H64R SOD1-GFP in the cells, and on its own, this protein remained diffuse throughout cytosol rather than aggregating. Only when the cells were exposed to the recombinant H64R SOD1 from Sf9 cells, aggregated with TFE, did they produce aggregates. In addition, when cells were infected with aggregates labeled with different colors and then co-cultured, they would exchange aggregates until both groups of cells contained aggregates of both colors; and fluorescent aggregates in culture media from one group of cells could be taken up into the cytosol of other cells. Thus, in a variety of paradigms (including without cell-cell contact), SOD1 aggregates can move from cell to cell and can seed soluble SOD1 to aggregate.
That last round of experiments was done with H64R SOD1 being expressed by all of the different cells, so they did not look at cross-seeding between mutant and WT SOD1.
Mutant Copper-Zinc Superoxide Dismutase (SOD1) Induces Protein Secretion Pathway Alterations and Exosome Release in Astrocytes [Basso 2013]
In this study they harvested cortical tissue from mouse embryos overexpressing mutant (G93A) or WT human SOD1, and isolated/differentiated astrocytes, then performed a number of tests on the astrocytes, including:
- Western blot on GFAP as a control - the GFAP band was a broader smear in the mutant SOD1 astrocytes, which they attribute to GFAP fragmentation due to calpain activation.
- 2D gels on pools of mutant SOD1 astrocytes (n=3) versus pools of WT SOD1 astrocytes and image analysis to identify dots that were more intense one gel than the other. They then identified these proteins by MALDI-TOF mass spec and technically validated the difference in protein abundance of a few of them with Western blots.
- Measurements of total protein secretion and exosome production.
- Exposing cultured motor neurons to exosomes from the astrocytes. They found that SOD1 was able to move from the exosomes into the neurons, and that the exosomes from the mutant SOD1 astrocytes caused some (36%) motor neuron death.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.