Targeting glutamate receptors in prion disease
A recent paper from the Aguzzi lab explores the possibility of targeting a class of neurotransmitter receptors — group I metabotropic glutamate receptors — as a therapy for prion disease [Goniotaki & Lakkaraju 2017]. The purpose of this blog post is to provide a review and critique of this work, but first, some biological and historical context.
What is a metabotropic glutamate receptor? Glutamate, that most delicious of amino acids, is a predominant excitatory neurotransmitter in the brain. Glutamate can bind to so-called ionotropic receptors such as NMDA and AMPA receptors, causing them to directly form a pore that allows Na+ ions to flow into the cell and K+ ions to flow out of the cell. Glutamate also binds to so-called metabotropic receptors, that term meaning simply that the receptors act through a second messenger system rather than directly forming an ion channel. This study focused on the group I metabotropic glutamate receptors, of which there are two types — mGluR1 and mGluR5 — both of which act as G protein-coupled receptors (GPCRs).
All of which brings back a flood of memories from introductory neurobiology: neurotransmitters and synapses and depolarization and potentiation and so on, that I don’t routinely think about in the context of this protein-folding disease. But this new study on the mGluRs is not totally out of the blue. There has been a small body of literature over the past few years examining possible interactions between PrP and glutamate receptors. For instance, there have been reports of PrP directly or indirectly affecting the activity of NMDA receptors [You 2012, Senatore 2012]. And there have been reports of mGluR5 being involved in a signaling pathway along with PrP and Fyn [Um 2012, Um 2013, Haas 2016] — you may remember this from Prion2015 if you were there.
But it’s not only the prion community that’s been talking about mGluR5. Indeed, it’s probably no coincidence that so many studies are now looking for roles of mGluR5 in incurable brain diseases. Part of the motivation may be because there has been a significant drug development effort centered on mGluR5 as a target. My first exposure to this was through the Fragile X community. Fragile X syndrome is caused by loss of function of the gene FMR1, usually due to a CCG repeat expansion in the promoter region that silences expression. One finding from the Fragile X knockout mouse was that it exhibited too much long-term depression or LTD — the process where receptors get less excitable the more they’re activated, the opposite of long-term potentiation. This excess of LTD was found to be due to an excess of mGluR activity [Huber 2002]. This led to the so-called “mGluR theory of Fragile X” [Bear 2004] — the idea that perhaps excessive mGluR activation was a critical central event mediating the pathogenesis of Fragile X.
Novartis developed an inhibitor of mGluR5, named mavoglurant (formerly AFQ056) and, promisingly, it was shown to correct this long-term depression problem in the Fragile X mice [Levenga 2011]. Mavoglurant soon went into clinical trials in Fragile X patients, but despite high hopes, the overall analysis didn’t show any evidnece that patients had benefitted. Some parents felt that their child had improved on the drug, though, and the data were suggestive that maybe the drug had only benefitted those patients with the most severe form of the genetic defect — a “fully methylated full mutation” resulting in a total loss of FMR1 expression [Jacquemont 2011]. In the hope that maybe this post-hoc analysis would replicate in a larger cohort, mavoglurant was then taken into further trials with a pre-specified stratification analysis on mutation methylation status. But here too, sadly, efficacy was not observed [Berry-Kravis & Des Portes 2016].
By which I mean, no one disputes that mavoglurant was pharmacologically effective — it definitely inhibits mGluR5 — but it didn’t lead to improved outcomes for patients in those trials. The authors of that study discussed several possible reasons why the trials may have failed, including patient age and treatment duration, but also the possibility that maybe the mGluR theory of Fragile X wasn’t correct, or at least wasn’t a big enough piece of the full story after all. In other words, maybe even though mavoglurant succeeded in targeting mGluR5, maybe mGluR5 wasn’t actually the right target in Fragile X. I myself don’t follow the Fragile X literature enough to have an opinion one way or the other on whether mGluR5 is the right drug target in that condition, and I know that many of those who are in the know believe the jury is still out. But I mention this because the question of “did we have the right drug target for the right disease?” is a crucial one, and is very often the reason why clinical trials fail [Bunnage 2011].
Moving on to the new study [Goniotaki & Lakkaraju 2017], the hypothesis was that mGluR1 and/or mGluR5 mediate some of the neurotoxicity of prions, and that inhibiting these receptors would reduce the neurotoxicity of prions, without affecting prion replication. The study used three compounds, shown below. note that each of the three is reported as being specific, with one only inhibiting mGluR1 and the other two only inhibiting mGluR5 [Gasparini 1999, Kohara 2008, Levenga 2011, Vranesic 2014]; none is reported to inhibit both receptors.
The first part of the study used cerebellar organotypic cultured slices (COCS) — primary cultures made from neonate mice overexpressing PrP. When these were infected with prions, each of the three compounds reduced the death of cerebellar granule cells at some particular timepoint when the slices were imaged (as far as I can tell, the paper doesn’t say what timepoint this was). The compounds also reduced the number of these cells in slices that died in response to toxic antibodies, relevant if you’re a believer in the N terminal toxicity hypothesis. And there are also a variety of other experiments regarding physical interactions of mGluR1/5 and PrP, and localization of mGluR5 and PrP within neurons.
But of greatest interest to me were the data from mice treated with MPEP, one of the mGluR5 inhibitors. The mice were fed MPEP in their food continuously from the moment of prion infection. The study found that prion-infected mice treated with MPEP declined slightly slower in terms of their performance on the rotarod — a standard behavioral test to see how long mice can stay balanced on a rotating bar. So for a few timepoints after the onset of symptoms, though the MPEP-treated mice were still impaired relative to their former selves, they performed better than the control mice. The study also found an extremely small but apparently statistically significant difference in survival between the two groups (+3-4%, P = 0.0008 or 0.02). Here’s the figure (note broken axes in C & D):
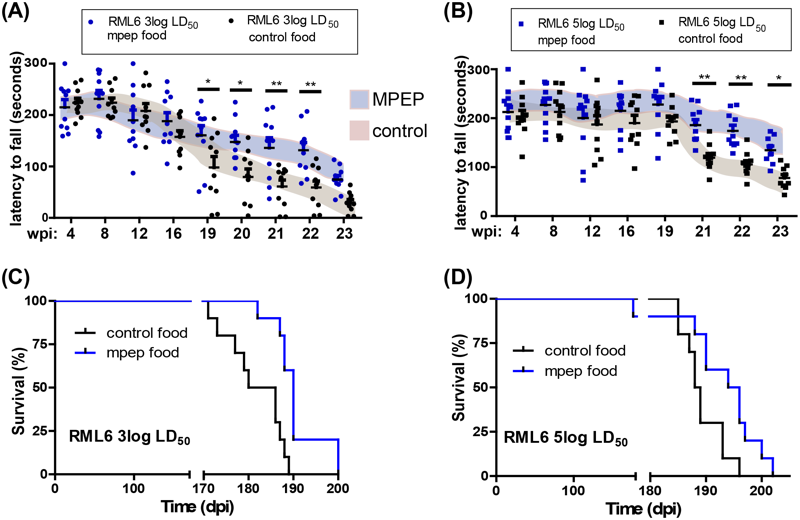
Figure 2 from [Goniotaki & Lakkaraju 2017]
The authors didn’t put a lot of emphasis on this tiny difference in observed survival, but instead focused on the idea that symptoms had been alleviated to a degree: the conclusion states that “mGluR5 antagonists may be useful for enhancing the quality of life of prion patients”. In other words, they are not claiming it’s a disease-modifying treatment that fundamentally alters the course of the disease, only that it might be used to manage symptoms.
I appreciate that framing, and it’s worth saying a bit more about symptomatic vs. disease-modifying treatment. There are drugs FDA-approved to manage the symptoms of other neurodegenerative diseases: memantine for Alzheimer’s, riluzole for ALS, tetrabenazine for Huntington’s. Pharmacologically, all of them modulate neurotransmitters in some way, tamping down excessive neuronal activation — in fact, memantine actually is a glutamate receptor (NMDA) antagonist. Yet we still think of these diseases as incurable, unstoppable neurodegenerative diseases. So even if mGluR inhibitors ever became an approved treatment for prion disease, it wouldn’t change my life or quest in any way.
But I think it’s unlikely that mGluR inhibitors will ever become a drug for prion disease, for a two reasons.
The first reason is timing. There is a mismatch in this study between the drug’s mechanism of action and the timing of the drug’s administration. The mice were given MPEP on a preventive basis, months before developing any symptoms. If the study is right that MPEP can slow down the process of neurons dying due to prion toxicity, then this early treatment may have been essential for observing any benefit. Yet what clinical scenario does this early treatment correspond to? No one is claiming that mGluR inhibition delays the formation or spread of prions, so it wouldn’t make sense for healthy people harboring PRNP mutations to take an mGluR inhibitor for years and years, just to have slightly alleviated symptoms for the few months they’ll be sick. The more likely clinical scenario is that mGluR inhibitors would be tested in a clinical trial in sporadic CJD patients, with some cognitive or executive function score as the trial endpoint, and if successful, the drug would be prescribed to patients after diagnosis. To predict whether an mGluR inhibitor would still be useful at that timepoint, it would be more informative to treat mice only after the onset of symptoms, which wasn’t done in this study.
The second reason is that question of do we have the right drug target? The motivation for this study was the idea that mGluR1/5 are involved in a pathway that transduces the neurotoxicity of prions. But the evidence for this ends up being mixed.
There is evidence, from this study and others [Um 2013, Hu 2014, Goniotaki & Lakkaraju 2017], that PrP interacts physically with mGluR5. But that’s also true for NCAMs and many other proteins that no one believes are involved in prion toxicity, and an unbiased, proteome-wide search for PrP interactors didn’t turn up mGluR5 as a hit [Schmitt-Ulms 2001, Rutishauser 2009]. Those earlier studies also reported functional evidence mGluR5 working with PrP to transduce toxicity, but they did so in the context of Aβ toxicity, and the relevance of PrP to Aβ toxicity in vivo has been called into question [Calella 2010].
The new study also cites a genome-wide association study (GWAS) in humans that turned up a variant in GRM8, the gene encoding mGluR8, as a possible hit [Sanchez-Juan 2015]. But there are a few reasons for caution here. First, the gene in that study was GRM8 on chromosome 7, not GRM5 which is in a completely different location on chromosome 11, and MPEP, the compound used for the in vivo experiments in this study, was found not to inhibit mGluR8, even at concentrations as high as 100 μM [Gasparini 1999]. Second, GWAS hits only provide evidence of locus linkage; the causal gene is not necessarily the one where the hit is found. For example, the gene FTO got its name from “fat mass and obesity associated” because an intronic variant in this gene is strongly associated with obesity, but it eventually turned out that the variant in question actually regulates IRX3 and IRX5, having nothing to do with FTO at all [Claussnitzer 2015]. Third, the GRM8 variant in that study was right at the edge of genome-wide significance (P = 1.7e-8 in one meta-analysis and P = 3.9e-5 in another meta-analysis), so while the association may prove to be real, I’d like to see further replication before being sure of it.
Then there is the animal model evidence. In the new study they obtained mGluR5 (Grm5) knockout mice and infected homozygous, heterozygous, or wild-type mice with prions to compare, and found no difference in prion disease between these groups [Goniotaki & Lakkaraju 2017]. This replicates the findings of another recent study [Beraldo 2016]. It would be surprising if inhibiting mGluR5 suppresses prion neurotoxicity, yet knocking it out entirely doesn’t. The study’s explanation for this discrepancy is that mGluR1 appears to be upregulated in mGluR5 knockout mice, though the evidence for this (Fig S4) was marginal (P < 0.05 in some brain regions at some timepoints but not others). They also found that slices from the brains of mGluR5 knockout mice did have reduced neuronal death in culture — but if knockout makes a difference in culture, why doesn’t it make a difference in the live mouse? If the results are not consistent between different experimental paradigms within the mouse, I would worry about the probability of successfully translating results from mouse studies to human clinical trials.
Probably the strongest evidence I’ve seen for mGluR5 involvement in prion disease is the rescue of neuronal loss that was seen in the cultured brain slices treated with mGluR5 inhibitors [Goniotaki & Lakkaraju 2017, Figure 1], which is fairly dramatic. But as noted above regarding the Grm5 knockouts, rescue in the slice model doesn’t always predict an effect in live animals. With the genetic knockout arguing against a major role for mGluR5, and a lack of human genetic evidence pointing to this gene in particular, the overall level of validation of mGluR5 as a target in prion disease starts to look pretty shaky.
I wanted to write a bit about this study because lately I have been spending a lot of time thinking about the incredible challenge of taking a therapeutic concept from preclinical studies to clinical trials. The prior is low — most clinical trials fail — so for me, the evidence that something will work has got to be very strong. This paper was an interesting case study. It’s tempting to want to bypass the years or decades of work that go into drug development and test a compound off the shelf that would be ready to go into clinical trials in an instant. And if something like that worked, it would indeed be amazing. But these types of studies have a low prior. Because no one has developed a drug for prion disease before, the drugs on the shelf aren’t going to target PrP, for which there is overwhelming evidence that it’s a valid drug target. Instead, they’re going to target some other protein with far more limited or mixed evidence for relevance to the disease process. In this case, the evidence supporting the drug target is not conclusive, the potential is probably limited to symptom management rather than prevention or reversal of disease, and the therapeutic effect measured in this one mouse model was rather limited. For all of these reasons, I’ll be surprised if mGluR inhibition ever makes it to a clinical trial. But I applaud the Aguzzi lab for taking a close look at this possibility, and for publishing all of these results, even the parts that were ambiguous or complicated.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.