FDA approval of the first RNAi drug

The headquarters of Alnylam, a few blocks from our lab in Cambridge, MA.
Late Friday afternoon, FDA announced the approval of patisiran, the first-ever drug based on RNA interference (RNAi). The drug is made of RNA targeting the TTR gene and will now be prescribed to people who have a mutation in that gene that causes a disease called hereditary transthyretin amyloidosis. As FDA Commissioner Scott Gottlieb put it:
This approval is part of a broader wave of advances that allow us to treat disease by actually targeting the root cause, enabling us to arrest or reverse a condition, rather than only being able to slow its progression or treat its symptoms… New technologies like RNA inhibitors, that alter the genetic drivers of a disease, have the potential to transform medicine, so we can better confront and even cure debilitating illnesses.
It’s undoubtedly exciting news, both for patients with that disease and for scientists who have been eager to see this new type of drug come to market. And it’s encouraging for us to see, particularly because the approach here is in many ways parallel to what we want to do for prion disease. But it is not quite the same approach, and Friday’s news doesn’t have any immediate impact on our path forward.
First, some background. The gene TTR encodes a protein called transthyretin, which is produced in the liver and secreted into the bloodstream and normally forms a tetramer, meaning four copies of the protein float around as a unit. Its normal job seems pretty dispensable but seems to involve transporting retinol (vitamin A) through association with another protein, retinol-binding protein [Monaco 1995]. In mice and some other mammals it also transports thyroid hormone [Episkopou 1993] though it doesn’t seem to do much of this in humans. This protein has the troublesome property that the four copies of the protein can dissociate from each other, whereupon each can become unstable. The protein then misfolds and aggregates into amyloid fibrils deposited on the peripheral nerves and in the heart. This can be caused by genetic mutations in TTR, most often the V30M mutation, in which case it can be called familial amyloid polyneuropathy (FAP) or, more precisely, hereditary transthyretin amyloidosis [Ando 2005]. The disease also has a sporadic, usually later-onset, form known as senile systemic amyloidosis (SSA) where people with no mutation develop the same plaques formed of normal, non-mutant transthyretin protein [Westermark 1990].
If the above description sounds familiar in some ways, yes, the disease has a lot of analogies to prion disease. In both cases, we have a problem protein whose normal function is non-essential but which can misfold either due to genetic mutations or just randomly. Accordingly, two of the same therapeutic strategies arise: bind and stabilize the protein to prevent misfolding, or reduce the amount of the protein altogether. But transthyretin amyloidosis has two advantages that explain why it’s years ahead of prion disease in the drug development pipeline. First, although transthyretin protein deposits on the nerves and in the heart, it is produced in the liver, which is an easier tissue to reach than brain. Indeed, liver transplantation is one option reported to improve health in people with TTR mutations [Holmgren 1993, Okamoto 2009]. Second, whereas prion protein (PrP) is a small, partly unstructured protein with no obvious binding pockets you can use to stabilize it, transthyretin has this thyroid hormone-binding pocket, an open invitation to develop a small molecule drug. Accordingly, for stabilizing the protein, there is tafamidis, a small molecule drug that binds in the thyroid hormone-binding pocket of the transthyretin tetramer. This keeps it from dissociating into monomers, which in turn makes it less prone to misfold. Tafamidis was approved in Europe in 2011 but FDA wanted to see more data before approving it in the U.S.; Pfizer announced positive Phase III data a few months ago.
A number of therapies to reduce the amount of transthyretin protein have been in the pipeline for years. This is a good place to introduce the difference between antisense oligonucleotides (ASOs) and short interfering RNA (siRNA). Both are short nucleic acid sequences designed to be reverse complement (A pairs with T, C with G) to a sequence in an RNA molecule of interest, in order to bind that RNA and cause its degradation. But they do so through two different cellular mechanisms, as shown below.
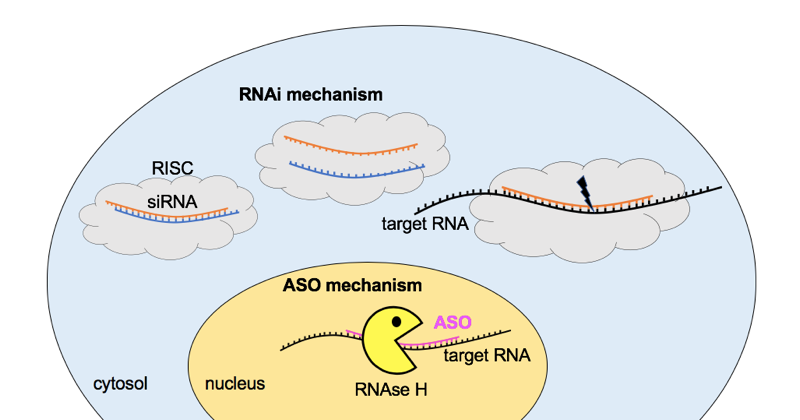
Comparison of the RNA interference (RNAi) and antisense oligonucleotide (ASO) pathways for RNA knockdown. For RNAi, short interfering RNA (siRNA) molecules consisting of a double-stranded complex of two 21-nucleotide strands overlapping by 19 nucleotides (see for instance the sequence of patisiran) are delivered to the cytosol, where they are taken up by the RNA-induced silencing complex (RISC), which uses one strand to recognize and cleave a target RNA. ASOs are single-stranded, chemically modified, ~20 nucleotide DNA molecules that reach the nucleus, where they bind a target RNA and cause the enzyme RNAse H to come and cleave it. Note that there are many simplifications here, and this is just one of the many mechanisms of action of ASOs.
In addition to the molecular mechanisms for RNA targeting being different, RNAi and ASOs differ in many practical respects of how the drugs are designed, formulated, and dosed. Among them, ASOs have the DNA backbone and/or sugars chemically modified in one or more ways that allow them to be directly taken up into tissue. Such backbone modifications are not often used in siRNA, which instead require for delivery either some sort of conjugation (attachment to a targeting molecule) [Nair 2014], encapsulation (in a lipid droplet that can enter cells) [Semple 2010, Tam 2013], or vectoring (the RNA is encoded in a virus that is delivered to infect cells) [Davidson & McCray 2011].
Both of these approaches, RNAi and ASOs, can achieve highly potent and specific reduction in one target RNA and thus in one target protein. Both have been in development for transthyretin for years. An antisense oligonucleotide against TTR, inotersen, developed by Ionis Pharmaceuticals and Akcea Therapeutics, just had positive Phase III data published [Benson 2018], received approval in Europe last month and is currently awaiting an FDA decision. Meanwhile, Alnylam had developed two RNAi drugs against TTR, the now-defunct revusiran and the just-approved patisiran. These two differed in their delivery strategy [Coelho 2013, Butler 2016]. Revusiran was chemically conjugated to a sugar called N-acetylgalactosamine (GalNAc), whereas patisiran is encapsulated in a nanoparticle made of a synthetic lipid called DLin-MC3-DMA [Jayaraman 2012]. Revusiran failed in a Phase III trial, with a disturbingly unexplained higher mortality rate in the drug-treated group than in the placebo-treated group [Garber 2016]. Alnylam promptly scrapped that candidate, and it was unclear whether there were negative implications for RNAi drugs generally. But then in late 2017, Alnylam announced positive Phase III results for patisiran, and many observers started to expect that approval was on the way. The full Phase III data published last month [Adams 2018] further reinforced that expectation.
As of Friday, FDA approval has now arrived, making patisiran the first RNAi drug in the U.S. This is a big milestone that people have been waiting for ever since the discovery of RNA interference 20 years ago [Fire 1998]. It’s great news for patients with transthyretin amyloidosis, and hopefully it will spur investment into new drugs with this mechanism for other diseases. As Commissioner Gottlieb explained at top, the approval of this RNAi drug is one example — along with a number of ASOs and other drugs — of how new drug modalities are targeting the underlying cause of disease and not just treating the symptoms.
All that being said, this news doesn’t change anything about our strategy in prion disease. As Sonia announced last month, we are working with Ionis Pharmaceuticals in an effort to develop an ASO drug for prion disease, one that would be delivered into the spinal fluid via lumbar puncture and reduce the amount of prion protein (PrP) in the brain. That’s still the right strategy because, as explained here, there are good proofs of concept for ASO delivery to the brain, both from an approved ASO drug (nusinersen for spinal muscular atrophy) and from ongoing studies in non-human primates and clinical trials in humans for a number of other ASOs.
While RNAi could in principle apply to prion disease, the big challenge is delivery. Whereas ASOs injected into CSF distribute broadly across the brain, as of today there just isn’t any good way to get siRNA into a large swathe of the brain. Alnylam’s pipeline doesn’t include any central nervous system targets, presumably for this reason. Companies that are working on RNAi therapies for the brain, such as Voyager are relying on injection of viral vectors directly into brain tissue, a strategy which only allows you to reach a small portion of the brain. See for instance their VY-HTT01 candidate for Huntington’s disease.
That approach has been tried in prion disease mouse models, and the results haven’t been too promising. The first study used a single injection of an RNAi lentiviral vector into the mouse brain, and while it was nominally reported as a positive result, the details didn’t look too promising [White 2008]. The effect of the treatment was mostly confined to the hippocampus, the brain region where it was injected, with only modest effects elsewhere in the brain, and any overall survival benefit was marginal. A recent study with a different viral vector (AAV2) found similar results. That vector was injected into a different brain region, the thalamus, and it reduced prion accumulation focally around the injection site but didn’t have much effect elsewhere in the brain and didn’t improve survival [Ahn 2014]. And bear in mind, both of these studies are in mice, and when you’re talking about diffusion of viruses through brain tissue, size matters. A mouse brain weighs about 400 mg, a human brain is about 1.3 kg — three thousand times larger. So for a focally delivered therapy injected directly into brain tissue, even if you could efficiently reach the entire mouse brain and dramatically delay disease in a mouse, it’s not clear whether that would ever translate to any benefit in a human. And if you can’t even achieve a substantial survival benefit in a mouse, which seems to be the status quo, then you’re a long ways from being able to do anything useful in a human.
Two animal studies achieved more substantial reductions in prion protein expression in the brain, but they did so by introducing the virus encoding the siRNA into the animal long before it was born, either through embryonic stem cells or through cloning [Pfeifer 2006, Golding 2006]. That’s a proof of principle that siRNA could work if delivered broadly, but it’s not a model of anything you could do in a human as a therapeutic. And some studies have also looked at other approaches to siRNA delivery to the mouse brain for prion disease. Instead of viral vector injections into the brain, two studies have used lipid nanoparticle formulations of siRNA delivered intravenously or rectally. But that didn’t seem very effective either — neither study reported a survival benefit [Pulford 2010, Lehmann & Relano-Gines 2014].
For now, ASOs are far and away the more likely drug candidate for prion disease, again for the reasons explained here. And while it’s exciting that a first-in-class drug was approved on Friday, ASOs are an established therapeutic modality in humans — the first ASO drug was approved in 1998, the year that RNAi was discovered. Still, I think the advent of RNAi drugs is good news. Perhaps it will stimulate more investment into new delivery methods, and maybe one day RNAi will become a viable approach for reaching the whole brain. We’re keeping our eyes on it and will be interested to see what develops.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.