Measuring PrP in spinal fluid two different ways
This week, Sonia and I are proud to announce two studies on quantification of prion protein (PrP) in cerebrospinal fluid (CSF): one study using ELISA, which has been on bioRxiv for a while and is now officially published [Vallabh 2019], and the other using mass spectrometry, newly posted to bioRxiv [Minikel & Kuhn 2019]. In this post, I’ll give a rundown of what you need to know about these studies. I’ll begin with why this topic is so important and how it fits into our goal of developing a drug for prion disease. I’ll cover the background on what was known about CSF PrP up to now. I’ll introduce each study, examining in turn what questions we asked, how we went about answering them, and what answers we got. I’ll conclude with some reflections on what role CSF PrP quantification will play in drug development for prion disease going forward, and what still needs to be done.
why CSF PrP?
Prion protein (PrP) is the protein that causes prion disease by misfolding in the brain. As explained in my intro to antisense, there are excellent proofs of principle that if we could lower the amount of PrP produced in the brain, that would be a safe and effective therapeutic strategy against prion disease. That’s why we partnered with Ionis Pharmaceuticals to develop antisense oligonucleotides (ASOs) to lower PrP. That project is ongoing, with Ionis working on bringing an ASO for prion disease to clinical trials in the coming years. Most of Sonia’s and my work here at the Broad Institute is focused on paving the way for this therapeutic strategy to succeed.
A key element of success will be the ability to determine whether, and by how much, a drug lowers PrP. Of course, what we really care about is PrP levels in the brain itself, but we don’t currently have a good way to measure brain PrP level in people who are still living. Because cerebrospinal fluid (CSF) bathes the brain and is accessible through a lumbar puncture, it’s often a realistic way to get a readout of protein levels in the brain. For example, for various proteins involved in other neurodegenerative diseases, including huntingtin [Wild & Boggio 2015] and SOD1 [Winer 2013], measuring the protein in CSF has turned out to be a good window onto what’s going on in the brain. Often, such windows prove critically important for advancing a drug.
As an example, consider RG6042, which is the Ionis/Roche drug candidate ASO designed to lower huntingtin protein in the brain, now in trials as a therapy for Huntington’s disease. RG6042’s first human trial was a Phase 1/2 trial designed primarily to assess safety, but also to see how much it could lower mutant huntingtin — the bad actor in Huntington’s disease — in patients’ spinal fluid. After the trial concluded, they announced that the highest tested doses of RG6042 lowered mutant huntingtin by 40% in spinal fluid. The trial was not designed to assess whether patients taking RG6042 improved clinically — the trial was too short for that — but the 40% figure showed that at least the drug was doing what it was designed to do, and the results presumably informed the selection of the dose for the Phase 3 trial which has now begun. So: measuring a protein in CSF can tell you whether a drug is doing its job, and what dose is necessary in order for it to do its job best.
In fact, we think that CSF PrP might be even more important — as mentioned in Kelly Clancy’s WIRED article about us, we’ve met with scientists at the U.S. Food and Drug Administration about whether lowering CSF PrP could be surrogate endpoint under the Accelerated Approval program. That full story will be a topic for a future post, but for now, suffice it to say that such a plan would only make CSF PrP measurement even more important, as it could be a basis not only for determining if a drug is doing its job and designing further clinical trials, but could actually support provisional approval of a drug. Long story short, one way or the other, the ability to measure PrP in CSF looks like it will be an essential piece of our effort to develop a PrP-lowering drug.
what was known
In early 2015, shortly after we first met with Ionis Pharmaceuticals and kicked off our ASO collaboration, we started to understand that CSF PrP quantification was going to be important. We surveyed the landscape to see what was already known. The answer was that a lot was known — thankfully, other scientists had put loads of work into this area and there were already several published papers on CSF PrP. But up to now, no one had been thinking of CSF PrP quite the way we were starting to think of it, and so they had asked different questions.
Whereas we were starting to think of CSF PrP as a potential pharmacodynamic biomarker — a way to measure the effect of a drug — all the previous studies had investigated CSF PrP as a potential disease biomarker — a measurement of a patient’s disease state. What all studies to date have found is that PrP concentration is lower in the CSF of symptomatic prion disease patients than in the CSF of patients with other, non-prion disease diagnoses [Meyne 2009, Schmitz 2010, Torres 2012, Llorens 2013, Dorey 2015, Abu Rumeileh 2017, Villar-Pique & Schmitz 2018]. That may seem strange, since prion disease is associated with an accumulation of misfolded PrP in the brain. But no one was too surprised by this, because there is a precedent: in Alzheimer’s disease, while Aβ plaques accumulate in the brain, Aβ in CSF goes down. Many of these studies examined whether lowered CSF PrP was a good enough disease biomarker to serve as a diagnostic — could a measurement of low CSF PrP indicate that a patient had prion disease? The answer seems to be that low CSF PrP by itself isn’t such a good discriminator between prion and non-prion diagnoses, but that if it is combined with other markers such as CSF Aβ, together they can be pretty good [Abu Rumeileh 2017], although CSF PrP is still unlikely to be a front-line diagnostic given that RT-QuIC has now emerged as such a highly sensitive and specific diagnostic tool.
But when you think about CSF PrP as a potential pharmacodynamic biomarker, a different set of questions emerges that hadn’t been answered by the above studies. These new questions all revolve around one central question: can we measure CSF PrP to determine whether a drug lowered brain PrP? This question gives rise to several sub-questions. Is CSF PrP stable over time in one individual? The studies cited above had found tremendous variability in CSF PrP between individuals, with the highest PrP concentrations being over 100 times the lowest ones seen. If there were that much variability within one individual, it would be very difficult to measure, say, a 40% decrease in CSF PrP due to a drug. If, on the other hand, CSF PrP is stable within one individual but is very different between individuals, what explains those differences, and are we confident we can reliably measure CSF PrP in the people we want to measure it in? In general, proteins in CSF can come from blood or brain. Can we figure out the relative contributions of these two tissues to CSF PrP, and is the brain contribution high enough that a decrease in brain PrP will be measurable? Would we need to exclude samples with significant blood contamination? In general, under what conditions can CSF PrP be quantified reliably enough to support its use in a clinical trial?
As we set out to answer these questions, an important issue to keep in mind is, what is the patient population in whom CSF PrP would be measured in a trial? There are effectively two patient populations in prion disease: those diagnosed after onset of symptoms (all sporadic and some genetic patients), and those with predictive genetic testing before the onset of symptoms (some genetic patients). These are very different patient populations on several dimensions, one of which is how CSF PrP might behave, a subject I’ll return to below.
Quantifying CSF PrP by ELISA as a pharmacodynamic biomarker
In the first study [Vallabh 2019], which Sonia led and which several of our collaborators in the prion field contributed to (thanks!) we used the BetaPrion Human ELISA kit (photo below). It’s a commercially available plate-based assay for PrP quantification and is the same kit used by most of the prior studies cited above.

The BetaPrion Human ELISA kit.
We used this to ask the following questions:
- How well does the kit perform technically?
- What factors, meaning either patient variables or pre-analytical factors (how the samples are handled prior to analysis) affect CSF PrP concentration?
- Is CSF PrP coming from the brain?
- Is CSF PrP stable in one individual over time?
To answer these questions, we analyzed a whole lot of samples using the kit, we perturbed samples in several different ways, and we measured other potential variables of interest in CSF.
To assess the kit’s technical performance, we worked from the FDA’s Guidance on Bioanalytical Method Validation, using CSF samples from a variety of non-prion disease patients. We spiked purified recombinant PrP into CSF to see how well it was recovered, we ran samples at differnet dilutions to see if the assay had linear performance, we mixed high- and low-PrP CSF samples to see if the mixture proportions came out as expected, we ran the standard curve and certain samples in a high number of replicates to look at reproducibility, and so on and so on. Retrospectively, across all the 41 plates of various samples that we ran for this study, we looked at metrics like coefficient of variation for technical replicates within and between plates, and so on. Overall, the result was that the performance is great. We did note a couple of minor caveats — there are some small but measurable position effects across the plate, and larger between-plate variability than within-plate variability, so if you have two samples you need to compare it makes sense to measure them right next to each other on the same plate. But at the end of the day, the answer is yes, this kit is capable of precisely measuring PrP concentration.
This successful analytical validation of the ELISA kit meant that the wide variability in PrP concentration between CSF samples, which had been reported and which we were also observing, was not just due to technical irreproducibility of PrP measurement — it was a genuine property of the samples being plated. What variables affected CSF PrP concentration? We had access to matched prion and non-prion disease CSF samples from collaborators, and we found, as others had before us, that PrP was lowered in prion disease patients, but this effect explained only a minority of the variance — there was still quite a lot of variability within any one diagnostic group. We looked at different tubes of CSF collected serially from the same individual to ask whether lumbar-thoracic gradient — position along the spinal column — mattered, but that didn’t seem to make a difference. We didn’t see unmabiguous evidence that age or sex were correlated with CSF PrP.
But one thing we noticed was that the same CSF sample aliquotted into different volumes wound up with different apparent PrP concentrations. We began to suspect that the issue was simply that PrP sticks to plastic like crazy. Sonia did an epically thorough series of experiments in which she made a huge number of aliquots of the same CSF sample and then perturbed them in different ways, such as moving CSF from plastic tube to plastic tube a variable number of times. The result was that every time CSF touches a different plastic tube, about half of the PrP is lost. She found that it was equally bad, or worse, on different types of plastic or glass. When you’re analyzing samples that may have been freeze/thawed, aliquotted, and transferred a variable number of times in variable volumes, this could result in a lot of variability in observed PrP concentrations. While we don’t have detailed histories of how all the CSF samples we had analyzed were handled in order to ask whether plastic exposure was indeed the hidden variable that explained much of the variance we observed, we could at least say that differences in how samples were handled prior to analysis — pre-analytical variability, it’s called — could potentially be sufficient to explain much of the variation. Luckily, it turned out that adding a small amount of detergent almost completely abolished this problem. While adding detergent after the fact couldn’t fully rescue the variability in retrospectively analyzed samples, this appeared to be a good solution for future sample collection in, say, a clinical research study or a clinical trial.
When we give talks about our work, Sonia sometimes laughs and points out how boring this all is. People don’t generally want to do (or fund, or publish, or hear about) research on how to store and aliquot and transfer biological samples, and in a sense they are right — it isn’t exciting, fundamental science. And yet, problems like plastic adsorption are the stuff of which failed clinical trials are made. If you do a trial where your goal is to measure PrP to see if it goes down, and you are losing half of the protein every time the samples touch plastic, you’re never going to be able to see the signal through all the noise. As much as we all like to hear about the big science, sometimes the little science is life-saving.
We sought to determine whether CSF PrP was coming from brain, and we found a few lines of evidence that it is. First, when we measured PrP concentration in CSF samples, in blood samples, and in brain samples, we found tons of PrP in brain, very little in blood, and an intermediate amount in CSF. So just in terms of abundance, it had to be that the overwhelming majority of CSF PrP was brain-derived. In further support of this, spiking blood into CSF didn’t change the level of PrP we could measure. And finally, CSF hemoglobin concentration — a proxy for the degree of blood protein contamination — was not correlated with CSF PrP concentration.
Finally, across several different cohorts, we looked at serial CSF samples taken from the same individuals on different days to assess within-subject stability or test-retest reliability as it’s known. Perhaps not surprisingly, in cohorts where the different CSF samples from the same individual hadn’t been handled uniformly — they were stored in different volumes in different types of tubes and so on — we saw a lot of variability in one person’s CSF PrP measurement over time. But in one cohort of placebo-treated controls with non-prion cognitive impairment from a clinical trial in which samples had been handled very carefully to minimize pre-analytical variability, we saw very good test-retest reliability, with a mean coefficient of variation (mean CV) of 13%.
All told, the results were very encouraging for the idea that CSF PrP could serve as a pharmacodynamic biomarker. Sonia had shown that ELISA-based measurement of CSF PrP was technically sound and reproducible. CSF PrP was very sensitive to pre-analytical variables, particularly plastic exposure, but that this variability could be acceptably minimized by adding detergent to CSF. CSF PrP was coming from the brain, not blood. Provided that pre-analytical variability was minimized, one person’s CSF PrP level was stable over time. Put all these factors together, and you reach the conclusion that a drug that reduced brain PrP by, say, 40% in a clinical trial should result in a decrease in CSF PrP concentration that would be readily measurable even in just tens of people.
One important caveat, though, is which people. We had confirmed, as several studies before ours had found, that CSF PrP concentration drops in symptomatic disease. And in fact, a recent study indicates that CSF PrP continues to decline longitudinally after disease onset [Villar-Pique & Schmitz 2018]. These conclusions are based on ELISA measurement of CSF PrP, which has certain limitations that motivated us to also want to quantify CSF PrP by mass spectrometry, hence our next study below. But assuming it is real, the decline in CSF PrP in symptomatic disease hints that it might be hard to read out the effects of a PrP-lowering drug in symptomatic patients by simply measuring CSF PrP. Would you expect that CSF PrP should go down after drug administration (because the drug makes it go down), or that it should go up (because the drug alleviates the disease process that is pushing CSF PrP concentration down)? This issue initially daunted us and made us unsure of how useful CSF PrP would be — that was the conclusion of my first blog post on this subject back in 2015. But Sonia’s study showed that even if CSF PrP could be hard to interpret as a pharmacodynamic biomarker in symptomatic prion disease patients, it could still be useful in pre-symptomatic people at risk for genetic prion disease, in whom we believe that the disease process, and any attendant decline in CSF PrP, has not yet begun. Of course, while all this is promising, there are still a lot of to-do items to make CSF PrP ready to serve as a biomarker in a clinical trial, and I’ll return to limitations and next steps at the end of this post.
Quantifying CSF PrP by targeted mass spectrometry

A mass spectrometer at the Broad Institute.
As the ELISA work above was ongoing, we were already at work on a second way to measure PrP using mass spectrometry [Minikel & Kuhn 2019].
From the prior literature and our own work with ELISA, we have seen again and again that CSF PrP is lower in symptomatic prion disease patients than in patients without prion disease. This poses a limitation for CSF PrP as a pharmacodynamic biomarker, because it means that you might only be able to get an interpretable readout of drug-induced changes in CSF PrP level in individuals in whom prion disease is not underway, such as pre-symptomatic individuals at risk for genetic prion disease. You might not be able to do a dose-finding study, for example, in symptomatic prion disease patients.
Why does CSF PrP decline in the symptomatic stage of disease? Prion disease is caused by a gain of function — meaning PrP is a bad actor protein that does something it shouldn’t do — and animal studies have shown that total PrP in the brain itself increases as prion disease progresses [Bueler 1994, Schulz-Schaeffer 2000, Moreno 2012], so the decline in CSF PrP seems paradoxical. It could be that, in disease, PrP is caught up in plaques in the brain instead of being shed into CSF, although the degree and type of plaque formation varies a lot between prion disease subtypes [Parchi 1999]. It could be that more PrP is intracellular — some studies have shown that whereas most normal PrP is on the cell surface, misfolding occurs in the endosomal-lysosomal pathway [Caughey 1991] and misfolded PrP may be preferentially diverted to intracellular compartments [Goold 2013]. It could be that PrP is downregulated as a result of the disease process, as animal experiments have shown that properly folded PrP in the brain decreases in the end stages of prion disease [Mays 2014].
Any of the above are plausible explanations, but we also considered another possibility. What if the CSF PrP is still there in symptomatic disease, but ELISA is just unable to detect it? ELISA depends on two antibodies, a capture antibody and a detection antibody, both binding the same copy of a protein. There was an effort to map the epitopes of the BetaPrion Human ELISA kit [Dorey 2015] but they only successfully mapped one of the two. It’s known that some epitopes of PrP are exposed in normal PrP but buried in misfolded PrP — this is the basis of conformation-dependent immunoassay [Safar 1998]. And it’s known that PrP can be chopped in half in a few different places (see alpha and beta cleavage and shedding), and that some of these cleavage events are increased in the disease state [reviewed in Altmeppen 2012]. So, we reasoned, there existed the possibility that some PrP in the CSF of symptomatic prion disease patients is simply invisible to ELISA because one of the epitopes is misfolded and inaccessible to antibodies, or because the protein has been cut between the two epitopes necessary for ELISA detection. If we could find a way to look at individual peptides of the protein, we might find that at least some of them are constant or even increasing in symptomatic prion disease. If so, that would afford us the ability to do dose-finding studies and get pharmacodynamic readouts for PrP-lowering drugs even in symptomatic patients.
There were other motivations for finding another way to measure CSF PrP as well. As a general matter, if your life depends on being able to precisely and reproducibly measure one analyte, you might want to have more than one good way of measuring it! This gives you some backup in case one option, say, becomes unavailable, or fails to meet some key criterion that regulators expect. In terms of more specific reasons, we found that the BetaPrion Human ELISA kit only detects human PrP — it didn’t detect mouse, rat, or even monkey PrP, so while we had a potential clinical assay in hand, we didn’t have a good quantitative assay to use in preclinical development of a PrP-lowering drug. The fact that PrP is omnipresent — everyone produces it all the time — also poses some challenges. For instance, in Sonia’s ELISA study she observed that across all CSF samples we analyzed, CSF PrP concentration was correlated with CSF total protein concentration. This could be real biology or, we suspected, it might be just because other proteins could adhere to the plastic surface of tubes, protecting PrP from being lost to plastic. But it was also pointed out to us that some ELISA assays suffer from matrix interference, meaning that while they principally detect the one analyte of interest (here, PrP), they also get some background signal from other proteins found in the matrix (here, CSF). The more of those proteins there are, the more signal you see, so this could also explain why CSF PrP and CSF total protein were correlated. For some biomarkers, the way you rule out this possibility is by studying “blank” matrix, but in our case that would mean CSF without PrP in it — and we’ve never yet found a homozygous PrP knockout human! One alternative is to quantify PrP in CSF by a totally different method, such as mass spec, which incorporates amino acid sequence information, thus giving you confidence that what you are measuring is really PrP and not just matrix interference.
For all these reasons, we approached Steve Carr and Eric Kuhn of the Broad Institute’s Proteomics Platform and asked if they would collaborate with us on developing a targeted mass spectrometry assay for PrP. What we ended up designing together is a multiple reaction monitoring (MRM) assay, in which CSF is denatured with urea and then digested with trypsin, which cuts the protein into short peptides, and the PrP peptides are quantified relative to known amounts of a standard — purified recombinant protein or synthetic peptides, labeled with stable isotopes to distinguish them from the endogenous PrP present in CSF. We chose peptides from across PrP’s amino acid sequence, from the N terminus to the C terminus, so that if PrP was being cut in half by any proteolytic cleavage events, we’d be able to see what was happening to both halves. The choice of peptides is diagrammed in Figure 1A, below, and the design of the assay is diagrammed in Figure 1B, below.
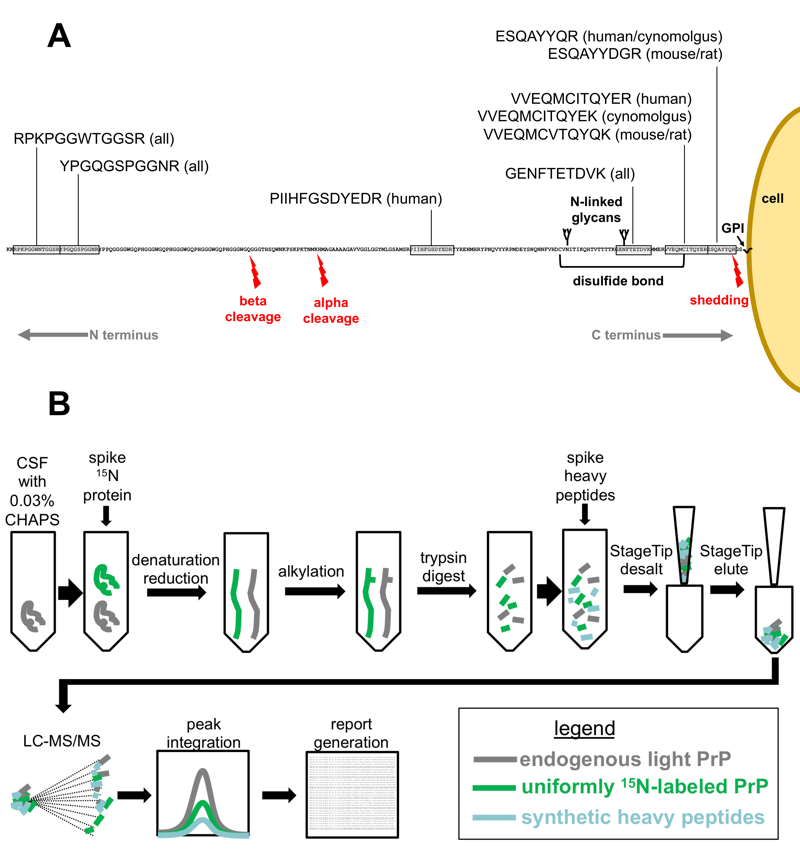
Figure 1 from [Minikel & Kuhn 2019]. A) Choice of peptides in the context of PrP and its proteolytic events, B) Assay workflow.
Eric Kuhn, who is deeply experienced in getting these sorts of assays up and running, was able to get a protocol working pretty quickly, and we started testing samples. First, we tested brain and CSF samples from a few different species to make sure the assay could detect what we thought it could detect, and we did some of the same sorts of analytical validation experiments that we had done for ELISA, to make sure that we could believe the data from the assay. The results looked good, so we proceeded to analyzing clinical samples — 55 of the same CSF samples from prion and non-prion disease patients that we had already analyzed by ELISA.
The main punchline is that when we compared the concentration of PrP, as measured by its tryptic peptides, in CSF between prion and non-prion disease patients, across the board, we saw exactly what had been seen by ELISA: PrP concentration is reduced in prion disease. The effect seemed to be about the same for all six peptides measured. Indeed, all six peptides were highly correlated with each other and with ELISA. So to a close approximation, at least, mass spec and ELISA seem to be measuring the same thing, or put differently PrP in CSF seems to be one thing.
The bad news is, this means that, sure enough, it might be really hard to do a dose-finding study of a PrP-lowering drug in symptomatic prion disease patients, where the level of CSF PrP really is on a downward trajectory. The good news is, CSF PrP seems to be a simple, well-behaved analyte. People studying tau in CSF have recognized that CSF tau is not just one thing — there are different isoforms of tau, there is phosphorylated and non-phosphorylated tau, and so on, and you can get very different answers depending on what you measure [Meredith 2013, Sato 2018]. If different parts of PrP had turned out to give different answers, that might raise questions that would complicate things, like which answer do we believe, and which one is important for knowing that you’ve lowered the thing you want to lower with a drug? The fact that PrP instead seems to be a simple analyte is a relief. Moreover, the fact that mass spec and ELISA seem to be measuring the same thing allows us to cross-validate the two methods with each other. For instance, we found that PrP concentration as measured by mass spec was correlated with CSF total protein concentration, just like we had found with ELISA — this increases confidence that ELISA really is specifically measuring PrP and is not subject to much in the way of matrix interference.
Going forward, it looks like either mass spec or ELISA could be suitable as a way to measure PrP and determine whether it is lowered in a clinical trial — and luckily, we now have both at our disposal.
next steps
Thanks to these studies, we now know a lot more than we did about CSF PrP and how it could function as a pharmacodynamic biomarker for a PrP-lowering drug. We think that, provided we have careful sample handling to minimize pre-analytical variabilty, we could readily measure a decrease in CSF PrP in response to a drug, as long as the trial is in pre-symptomatic people who are at risk for genetic prion disease but in whom the disease process has not yet begun. But there’s still a lot more work to do to make this biomarker usable in a clinical trial context. The two biggest tasks are as follows.
First, while all the evidence suggests that CSF PrP should respond to a decrease in brain PrP concentration upon administration of a PrP-lowering drug, we haven’t yet shown this empirically. Of course, showing this empirically in humans will have to wait for a clinical trial — that’s the whole point. But we still want to do experiments in animal models to satisfy ourselves that if we lower brain PrP this will indeed be reflected in a readout of CSF PrP.
Second, while we found good test-retest reliability of CSF PrP in the one cohort described above, those were patients with non-prion cognitive impairment — not the patient population we want to treat in a trial. We still need to establish that CSF PrP is stable over time, in the absence of drug treatment, in pre-symptomatic people at risk for genetic prion disease. This is the primary goal of our clinical research study at Mass General Hospital, which launched in 2017 and which is continuing to recruit.
Hopefully we’ll have updates on those areas of effort over the coming year or two. In the meantime, our studies that came out this week provide encouraging data, and we are optimistic that CSF PrP will prove useful as a biomarker. Have a read [Vallabh 2019, Minikel & Kuhn 2019], and if you have any thoughts, please feel free to leave us a comment below.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.