Is peripheral neuropathy in prion disease caused by a loss of PrP function?
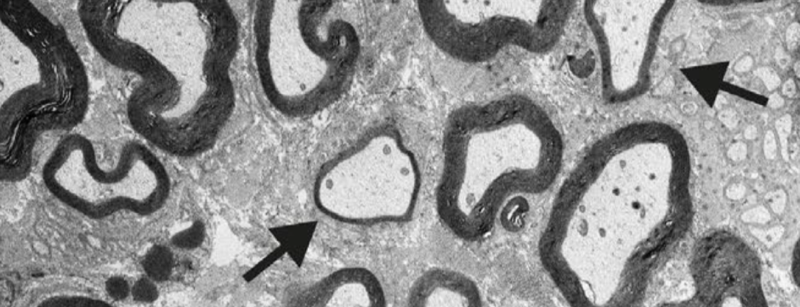
Myelin thinning in peripheral nerves in PrP knockout mice, from Figure 6E of [Nuvolone & Hermann 2016].
A number of studies have documented the prevalence of peripheral neuropathy — sensory and/or motor defects in peripheral nerves — as one among many presenting symptoms of prion disease in a subset of patients. Peripheral neuropathy is also the sole clearly documented phenotype associated with PrP knockout. This has led some researchers to speculate that prion disease, although clearly caused by a gain of function due to change in conformation of PrP, might also result in some loss of PrP function. Is the peripheral neuropathy in prion disease a causal result of loss of function, or are the two phenomena “true, true, unrelated”?
peripheral neuropathy associated with PrP knockout
Knockout of prion protein (PrP) seems to be well-tolerated. In humans, we have not yet seen a complete knockout, but we see no evidence of any phenotype in individuals with one inactivated allele, nor any evidence that natural selection is at work against loss-of-function mutations in PRNP [Minikel 2016, Minikel 2019]. Mice, cows, and goats lacking PrP seem to be outwardly normal [Bueler 1992, Richt 2007, Yu 2009, Benestad 2012] — dozens of knockout phenotypes have been claimed in the literature, but most have not been robustly documented and reproduced, and some have been outright refuted or attributed to genetic confunders [Steele 2007, Wulf 2017]. The one knockout phenotype that is inarguably real is a progressive demyelinating neuropathy seen only in peripheral nerves, not in the central nervous system, and only in complete knockouts, not heterozygotes [Bremer 2010]. It has validated across knockout mouse lines [Bremer 2010, Nuvolone & Hermann 2016] and in goats [Skedsmo & Malachin 2020] and has been attributed to the loss of agonistic activity of a PrP N-terminal signaling peptide on a G protein-coupled receptor, Adgrg6, expressed on Schwann cells [Kuffer & Lakkaraju 2016]. This defect in myelin maintenance is readily visible on histological sections of peripheral nerves, but phenotypically, it is mild to the point of being nearly silent. A difference in time to react to a hot plate was detected for homozygous knockout mice at age 60 weeks only sometimes, depending on the genetic background of the mice [Bremer 2010], and no outward difference at all was observed in knockout goats up to 7 years old [Skedsmo & Malachin 2020].
peripheral neuropathy in prion disease
The presenting symptoms of prion disease can be exceptionally diverse. All cases converge over time on profound dementia, but the earliest signs might be cognitive, or might be motor or psychiatric, or something else: weight loss, vision trouble, and headache can all be first symptoms [Will & Matthews 1984, Brown 1986, Rabinovici 2006]. Within this diversity of presentation, sensory changes have been recognized as an early symptom in 5-9% of cases [Will & Matthews 1984, Rabinovici 2006]. While “sensory” changes could originate from peripheral or central nervous system dysfunciton, there have long been a smattering of case reports where peripheral involvement has been specifically demonstrated through nerve conduction studies, nerve biopsy, or other tests, in both sporadic and genetic prion disease [Neufeld 1992, Antoine 1996, Esiri 1997].
In recent years, awareness of peripheral involvement in prion disease has grown considerably. A couple of years ago, prion seeding activity was detected in the skin of sporadic CJD patients [Orru 2017], and while the cell type of origin was not elucidated, one theory is that it’s coming from the peripheral nerves. And a study last year found that peripheral neuropathy may be more common in sporadic CJD than we thought: 31% (86/277) of patients had signs or symptoms of neuropathy such as pain, weakness, or numbness, and in the subset of those that underwent electrophysiological or histological studies, there was often clear evidence of peripheral nerve involvement [Baiardi 2019]. Peripheral signs were relatively more common in patients with a 129MV genotype, which is also associated with more slowly progressive disease. Still, it appears that the neuropathy in sporadic CJD, when present, is just one feature within an overall neurological catastrophe leading to death within a few months. Neuropathy can be broadly categorized into axonal (due to damage to or loss of the nerve axon) or demyelinating (due to loss of myelin), and it appears that the neuropathy in sporadic CJD can be either or mixed [Baiardi 2019].
Meanwhile, we’ve also come to understand better the spectrum of symptoms associated with PRNP truncating variants that cause prion disease. These are nonsense or frameshift mutations near the C terminus of the protein that leave most of PrP intact, while removing the GPI anchor, thus changing PrP from a cell surface protein to a secreted protein and causing a gain of function. Such mutations were first reported to cause an Alzheimer’s-like slowly progressive dementia [Kitamoto 1993], but we now understand that peripheral amyloidosis, leading to gastrointestinal and/or peripheral nerve involvement, can also be a pronounced feature [Mead & Gandhi 2013]. In patients with truncating variants, the neuropathy often progresses gradually for years or decades before any cognitive changes [Mead & Reilly 2015]. In people with truncating variants, the neuropathy has been reported as primarily axonal in nature [Mead & Gandhi 2013, Fong & Rojas 2017, Bommarito 2018, Capellari 2018].
One recent paper reported a prion disease patient compound heterozygous for two PRNP mutations, V210I and 2-OPRD (deletion of two octapeptide repeats), with initial symptoms of “dizziness and disequilibrium” [Piazza 2020]. This individual also had numbness in the arms and legs, and electrophysiological studies suggested demyelinating polyneuropathy. Neither of these mutations has evidence for being highly penetrant, but of the two, V210I seems the more likely cause of the patients’ disease, as it appears to confer lifetime risk roughly on the order of 10% [Minikel 2016]. 2-OPRD has been seen in just a handful of prion disease patients (3 out of 10,460 in a case series), never with a positive family history, and while our ability to call such large deletions in exome sequence data is still limited, this variant appears to have appreciable frequency in the general population (19 and 4 alleles for two DNA variants leading to this deletion in gnomAD).
neuropathy in peripheral amyloidoses
PRNP truncating variants represent a rather rare subtype of prion disease. But systemic amyloidosis associated with other proteins, besides PrP, is far more common, with some estimates saying it causes up to 1 in 2,000 deaths [Wechalekar 2016]. This class of diseases can be caused by a wide variety of different proteins that go wrong, misfolding and depositing as amyloid in a range of different tissues, leading different types of organ failure and ultimately death. How common is peripheral neuropathy in these diseases? From a quick literature review, it seems to depend a lot on the causal protein [Plante-Bordeneuve & Said 2011, Wechalekar 2016]. Some proteins, such as fibrinogen Aα-chain, build up in other tissues such as the kidney, and rarely, if ever, cause peripheral nerve problems. Others, such as transthyretin, almost always cause peripheral neuropathy:
| disease | causal protein (gene) | frequency of peripheral neuropathy | reference |
|---|---|---|---|
| wild-type or hereditary TTR amyloidosis; familial amyloid polyneuropathy | transthyretin (TTR) | ~100%, varying severity | Hund 2001, Mariani 2015 |
| gelsolin amyloidosis | gelsolin (GSN) | “most” kindreds | Kiuru-Enari & Haltia 2013 |
| AL amyloidosis | IgG light chain (IGL/IGK) | 35% (51/146 cases) | Duston 1989 |
| ApoA1 amyloidosis | ApoA1 (APOA1) | ~17% (2/12 families) | Joy 2003 |
| fibrinogen amyloidosis | fibrinogen Aα-chain (FGA) | ~0% (1/69) | Gillmore 2009 |
conclusion
At first glance, it appears there is a lot here that is too interesting to dismiss as coincidence. Peripheral neuropathy is the one clear phenotype of PrP knockout mice, and it’s also a not-uncommon early symptom in prion disease. What’s more, it’s been observed in patients heterozygous for PRNP truncating variants, where the prior for the mutation abolishing the protein’s native function seems high, as well as in a patient compound heterozygous for two different PRNP mutations, inviting speculation that the mutations could both interfere with native function, making the patient effectively null. To be clear, there’s no doubt that prion disease is caused by a gain of function, but just because a misfolded conformation and/or a mutation cause a new toxic function doesn’t exclude the possibility that they also abolish native function. For example, there is evidence that PrP with the pathogenic D178N mutation is constitutively under-expressed [Petersen 1996, Parchi 1998, Jackson 2009, Watts 2016, Villar-Pique & Schmitz 2018, Vallabh 2019], and there is evidence that PrP can be post-translationally downregulated in the brain as prion disease progresses [Mays 2014]. In each case, PrP is causing prion disease via a gain of function, but its lowered abundance might also somewhat compromise its native function along the way.
After reading more deeply, though, I believe the weight of the evidence suggests that the neuropathy in PrP knockout mice and the neuropathy in prion disease patients are true, true, unrelated. Here is my reasoning:
- Timing: The histological signs of peripheral neuropathy in PrP knockout mice progress gradually over a lifetime, whereas the neuropathy reported in sporadic CJD, E200K CJD, and the compound heterozygous V210I/2-OPRD patient, always set in suddenly in mid-life, roughly concurrent with the onset of other symptoms in the context of a rapidly progressive disease.
- Type: The peripheral neuropathy in PrP knockout mice is demyelinating in nature, whereas that in prion disease can be either demyelinating and/or axonal. Indeed, it is primarily axonal in patients with PRNP truncating mutations, who might otherwise serve as a counterpoint to my point #1 above, because their neuropathy is slowly progressive and precedes central nervous system involvement.
- Prior: Peripheral neuropathy is not uncommon in other peripheral amyloidoses caused by proteins such as transthyretin and IgG light chain, whose native functions have nothing to do with myelin maintenance. Therefore, the fact that peripheral prion propagation and prion systemic amyloidosis can cause peripheral neuropathy may not be such a coincidence at all, and may not need any explaining.
That’s certainly not the end of the story, and one could certainly still posit even more complex explanations for the observed data. We still don’t know the mechanism by which prions kill neurons in the brain, so we’re far from being able to speculate as to why they might also damage nerves in the periphery. I’m curious to hear what readers think, but for me, the Occam’s razor is that some sort of gain of function due to PrP misfolding is what damages the nerves in a subset of prion disease patients with peripheral neuropathy.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.