Huntington's disease ASO trial halted
the news
On Monday, Roche/Genentech announced the sad news that the Phase III trial of tominersen, a huntingtin (HTT)-lowering antisense oligonucleotide for Huntington’s disease (HD), was halted early. Ionis, which developed the drug candidate before licensing it to Roche, followed with its own press release. The announcement spawned a rash of news articles, but if you only read one, let it be yesterday’s HDBuzz post.
What happened? This was a double blind clinical trial, meaning neither the patients nor the doctors treating them know who is on drug and who is on placebo. In such cases, the typical practice, enshrined in FDA guidance, is that an indepednent data monitoring committee (iDMC) is charged with keeping tabs on the trial. This group of a handful of people (at least three), generally including physicians and biostatisticians. They review at pre-specified intervals all the unblinded data coming in from the trial, which even the companies themselves do not see, and if they see any reason the trial should not go on, they can call a premature halt. Occasionally, this can be good news: sometimes a drug is so obviously effective that an iDMC declares it would be unethical to continue a randomized trial. But often, it’s bad news: the data make an favorable risk/benefit balance unlikely, meaning either that the drug appears less safe than expected, and/or, very unlikely to prove effective.
In this case, the iDMC simply recommended that the Phase III trial should “discontinue dosing” based on “the investigational therapy’s potential benefit/risk profile for study participants.” They did say that “no new or emerging safety signals were identified,” but beyond that, no details were given. To be sure, it’s bad news, but we don’t yet know just how bad or what kind of bad. This update, cryptic as it is, had to be blasted out to the whole world simultaneously, to avoid a situation where some privileged individuals know first, and can engage in insider trading. As far as we can tell at present, with the exception of the handful of people on the iDMC itself, no one on Earth knows anything more than what I’ve just told you in this paragraph — perhaps not even people at Ionis, Roche, and Genentech, not even the neurologists who’ve been injecting the drug.
Why this blog post, then? My initial reaction was to hold off writing a post until we actually know anything, rather than engage in wild speculation. But enough people have emailed me in the past 24 hours that I decided it was important to acknowledge the news and provide some, limited as it may be, guidance to the prion disease community on what this might mean.
what it means for HD
For our friends who study HD, are at risk for or suffer from HD, or both, the news is devastating, though not, I will argue, the end of the world. While there are many trials ongoing in HD, tominersen had a strong therapeutic hypothesis — lowering the protein that causes the disease — and strong data from a Phase I trial, showing it could lower that protein by 40% [Tabrizi 2019]. There are several other therapies in development to lower huntingtin protein, but tominersen was way out ahead, and the discontinuation of this trial is a major setback and disappointment. How big a setback depends on what happened and why. There are several possible interpretations, and while the iDMC surely knows more than us, even they may not know why.
One possibility is safety. The press release stated that “no new or emerging safety signals were identified.” That pretty much rules out any dramatic adverse event like the lower limb weakness seen in an Angelman’s syndrome trial last year. It is not clear to me, as an outsider, whether this particular choice of wording is also intended to rule out a safety issue that was neither “new” nor “emerging” — say, if an adverse event identified in the Phase I was simply starting to prove a bit worse or a bit more common than anticipated. The Phase I trial did identify a transient increase in NfL (a marker of neuronal damage) after dosing initiation, but as discussed in a 2019 HDBuzz post, this eventually went away, and in the end, NfL rose less in the drug group than in the placebo group. In terms of side effects, the Phase I data were super clean, with adverse event rates about identical in the drug and placebo groups. Moreover, as noted in the Ionis statement, the iDMC did not recommend that dosing be halted in a separate, concurrent trial evaluating the pharmacology of tominersen. All this argues against a safety issue per se, but it’s hard to say anything for sure.
The other possibility is efficacy, though this term could mean a range of things. The Phase III trial launched in Jan 2019 and was slated to run through Mar 2022. The iDMC might have observed that HD symptoms were progressing at least as quickly in the drug group as in the placebo group, and have therefore concluded that the weight of the first two years of study data made it impossible or nearly impossible that the final year of data would tip the scales back to a positive outcome. But why? The HDBuzz post mentions three possibilities, all of which merit consideration:
- Disease stage - did we need to lower huntingtin earlier, perhaps before symptom onset, in order to make a difference?
- Potency - did we lower huntingtin either too much (it’s an essential gene) or not enough?
- Allele specificity - did we need to aim only for mutant huntingtin rather than (as tominersen did) both mutant and wild-type huntingtin?
The good news is that for each such possible explanation, there exists a pivot: do premanifest HD trials, test higher or lower doses or more potent drugs, develop allele-specific drugs. Many such efforts are already underway, it’s just that they’re a few years behind where tominersen was.
what it means for prion disease
For years now, we’ve been working with Ionis on an ASO to lower PRNP for prion disease. What does the halting of tominersen’s trial in HD mean for our efforts? It doesn’t mean much yet, because it is too early to say. Only a few people on Earth know what the iDMC saw, and even the iDMC may not know why they saw what they saw. Unpacking that result and figuring out what, if anything, it means for other ASOs in the pipeline will take a long time. Right now, a very wide range of possibilities remains open. The result in HD could turn out to mean something very specific to that compound, that patient population, or that target, and have few if any implications for ASOs for prion or any other neurological disease. Or it could turn out to tell us something about the pharmacology or tolerability of ASOs that is at least somewhat more generalizable, and could in turn range from “blip” to “major pivot” in terms of our strategy.
In the meantime, this does not mean that we or Ionis have given up on an ASO for prion disease! An unfortunate coincidence is that earlier this year, Ionis made a corporate decision to stop listing preclinical programs in its official pipeline — they decided that doing so gave away too much information to Ionis’s competitors, many of whom provide little or no information about their own preclinical programs. Therefore, as early as January, PRNP (along with dozens of other targets) was removed from Ionis’s website. That change pre-dates the tominersen news, and does not mean they’ve abandoned the program.
some perspective
As I said up top, right now we don’t know much of anything about what happened with tominersen. This whole post is just a long way of saying we don’t know anything.
But, many of you who are still reading this post are here because your family has prion disease, and that particular life experience can certainly cultivate a knack for imagining the worst case scenario.
So consider one such worst case scenario. You’re a patient with an incurable disease and high hopes for a new type of drug. The therapeutic hypothesis is rock solid — lowering the causal protein — and the drug gets to Phase III. Then, the sky falls: the iDMC stops the trial on finding that there have been 18 deaths in the drug group and just 2 in the placebo group. No one knows why these people died, but somehow, the unthinkable has happened: the drug is much, much worse than nothing. Is all hope lost? This all happened in 2016. The disease: transthyretin amyloidosis. The new drug modality: RNAi. The drug candidate: revusiran, which targets TTR. The sky fell, and to this day, no one knows for sure why more people died on drug [Judge 2020]. Alnylam’s stock lost 50% overnight. But it was all the dark before dawn. They pivoted. All it took was two years’ time and a different delivery mechanism (intravenous nanoparticle-encapsulated RNA rather than subcutaneous GalNAc-conjuagated RNA). In 2018, patisiran proved effective at lowering TTR and improving outcomes in transthyretin amyloidosis, and it became the first approved RNAi drug. And by the way, there’s now an approved ASO for TTR too: inotersen. A good therapeutic hypothesis is bigger than just one drug or one modality.
ASOs, for their part, have been around 30+ years, and have many times been declared dead on arrival, then hailed as a savior, then declared dead again. Look at Ionis’s stock price for the past 5 days, and thanks to the auto-scaled y-axis, you see a cliff at the end of the world:
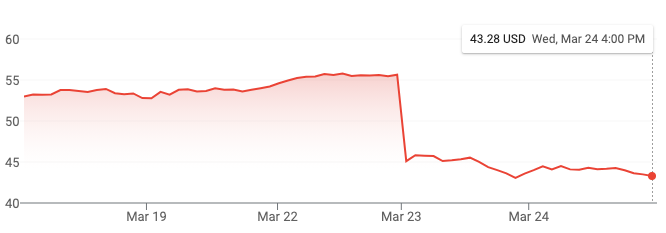
But zoom out to 5 years, and this is but one of many ups and downs:
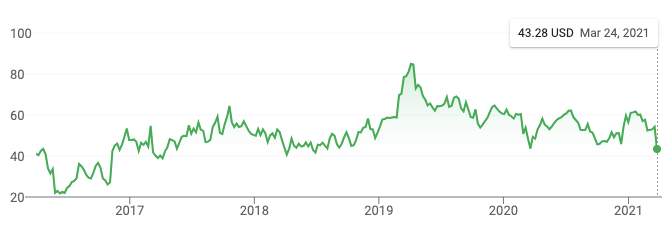
Of which, the first big up, in November 2016? That’s nusinersen proving effective in spinal muscular atrophy — don’t forget, we know that ASOs can be safe and effective in the CNS, and they are saving lives today.
I’m a Stockdale optimist: “You must never confuse faith that you will prevail in the end — which you can never afford to lose — with the discipline to confront the most brutal facts of your current reality, whatever they might be.” I take this week’s news seriously: I allow that it could turn out not to mean much for prion disease, and I also allow that it could be the beginning of some major obstacle for us. If so, tackling major obstacles is what we’ve done since day one, and it’s what we’ll keep doing.
For the nearly ten years of this quest so far, I have been fueled every day by ferocious, righteous optimism, while also acknowledging with open eyes the undeniably abysmal success rates in drug development. According to estimates over the last decade, only 8-14% of drug programs entering Phase I trials eventually lead to an approval [Hay 2014, Wong 2019, Thomas 2021]. True, it is fortunate that the success rate appears to double if your therapeutic hypothesis is rooted in human genetics [Nelson 2015], and it also appears to be higher for rare diseases [Thomas 2021], and those are both things we have going for us. Even still, if you take a cold, hard look at the numbers, even at the moment when the first human gets the first dose, it is hard to convince yourself you have better than even odds of the drug turning out to be both safe and effective. And we’re not even at Phase I yet — our program is still preclinical. People who spend their lives developing first-in-class therapies operate on a y-axis of difficulty whose scale most people in most industries cannot fathom. Making drugs is really, really hard. But you try, try again. I owe my life to modern medicine more than one time over, as I bet most of you do (if for no other reason than vaccines) — most of us are here because someone tried, and tried again.
So although it’s way too early, because we still know nothing, imagine for a moment that the worst is true, and the tominersen data ultimately reveal something terrible that scuttles the PRNP ASO program. If so, what did we get for all our effort over the past five years? A lot. And I mean that: pharmacologic validation of a therapeutic hypothesis, a pharmacodynamic biomarker to go with it, a clinical strategy for how to develop it, natural history data to support that clinical strategy, and much more. A disease that many would have called a lost cause or a charity case ten years ago, is now, in industry parlance, “developable”.
But for now, my suggestion is not to focus on any particular scenario. It will be a while before we know more, and in the meantime, there is so much work to do.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.