The role of different brain cell types in prion disease
As we think about developing a PrP-lowering drug, we think of prion disease as a whole-brain disease, not confined to any one brain region. But what cell types does a drug need to reach in order to A) be effective, and B) move a CSF PrP biomarker? I’ll start by unpacking where PrP is expressed at the RNA and then the protein level, and then dive into the role of different cell types in disease.
where is PrP expressed?
PrP RNA expression
In the early days of the prion hypothesis, it was considered a matter of fundamental science to determine when and where PrP RNA was being synthesized — was it in only infected animals or healthy ones too, did its transcription increase as prion disease progressed, and in what cell types was it produced? The answers to the first two questions were easy: yes, both infected and uninfected animals expressed PrP RNA at the same level, and its level did not change as a function of disease progression [Chesebro 1985], Kretzschmar 1986]. Expression by cell type proved tricker to reach a consensus on, even though the method — in situ hybridization (ISH), in which a radiolabeled antisense probe is used to locate an RNA molecule within tissues — has been around for ages. Two ISH studies found that in hamster [Kretzschmar 1986] and in chicken [Harris 1993], virtually all PrP RNA was in neurons, with negligible concentrations in glia or other cell types of the CNS. But other studies in mouse embryo [Manson 1992] and sheep [Hunter 1994], and both young and adult hamsters and rats [Moser 1995] found PrP RNA in astrocytes, oligodendrocytes, and epithelial cells. That answer seemed to align better with the evidence that PrPSc in terminally sick prion animals can be seen astrocytes [Diedrich 1991] as well as in white matter (where glia are present along with neuronal axons but not neuronal soma) Taraboulos 1992].
More sophisticated modern methods have found PrP RNA expression across all different cell types in the brain. Just before single cell sequencing took off, Ben Barres’s lab isolated and cultured different cell types from fresh mouse [Zhang 2014] and human [Zhang 2016] brain tissue by immunopanning — creating cell suspensions and then pulling down types of interest with ligands or antibodies. The transcriptomes of different cell types from these experiments can be browsed at brainrnaseq.org — here is the plot for PRNP:
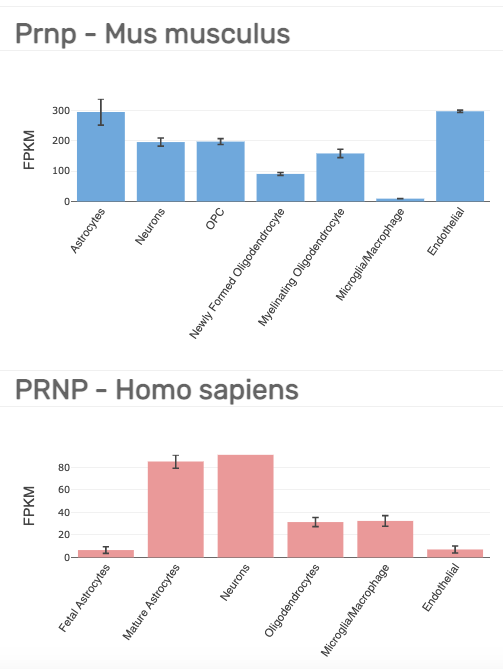
The lowest bar in either of the above plots, 6.5 FPKM, is still well above the noise: EBI uses 0.5 FPKM as a default minimum for an interpretable expression level. Thus, PRNP seems to be expressed ubiquitously across brain cell types, just as it is expressed ubiquitously across tissues. But, if you want to get more quantitative and ask which cell types express relatively more or less PRNP RNA, there is unfortunately not great agreement between the mouse and human experiments above. For instance, in mouse, endothelial is highest at 297 FPKM, while in human, it is near the bottom at 7 FPKM.
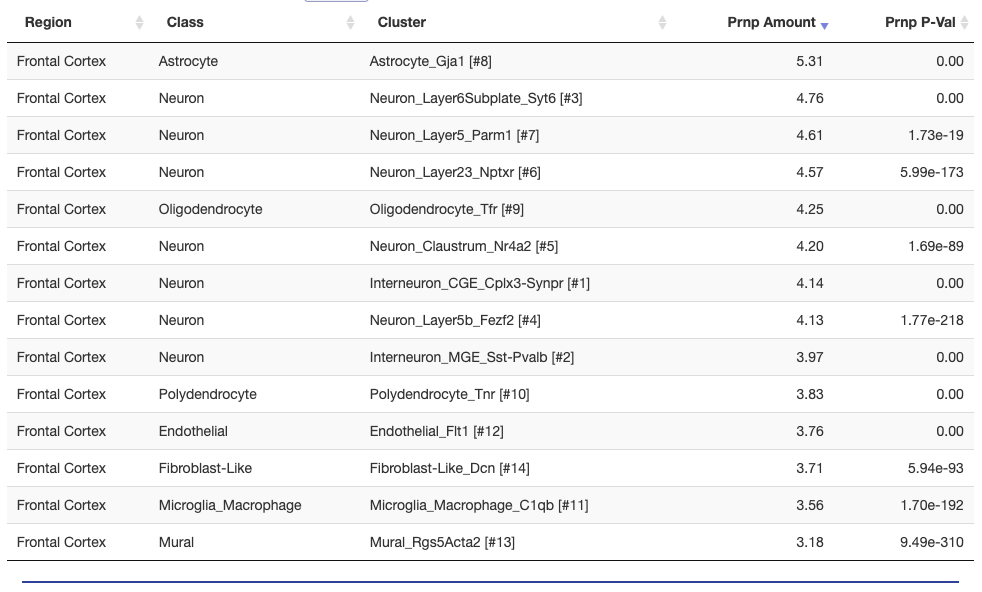
This theme — we agree that PRNP is expressed everywhere even if the relative differences between cell types are not consistent — is mirrored in more recent single cell datasets as well. Drop-seq [Macosko 2015, Saunders & Macosko 2018] single whole cell data hosted at dropviz.org show almost identical expression levels between all cell types in mouse cortex. Astrocytes narrowly eke out first place, while microglia and mural cells (smooth muscle and pericytes in the microvasculature) are last:
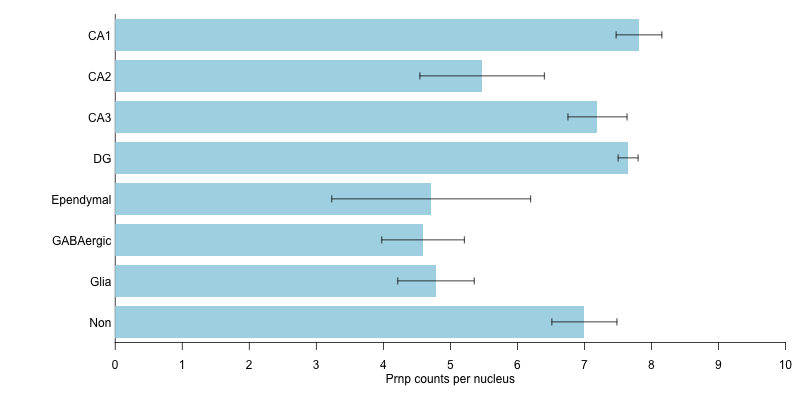
I also parsed out the data from DroNc-seq [Habib 2017], which is nuclei rather than whole cells, and a different method and approach. Again, all the cell types they clustered their data into fell within a pretty narrow range of Prnp expression levels:
Allen Brain Atlas has a large number of single cell datasets hosted in their browser, for example the human M1 10X browser shows the following. Cells are hierarchically clustered by type, so most of the plot’s width is taken up by different populations of neurons, while all the non-neuronal cell types are clustered at far right.
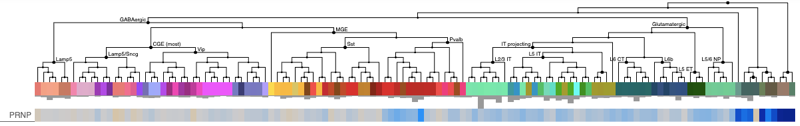
Here, the non-neuronal cell types (dark blue bars at far right) have trimmed mean expression ranging from 0-3, while the neuronal cell types are mostly in the 6-8 range (beige and light blue across most of the plot). The picture from Allen’s human cortex Smart-seq is nearly identical — again, see that dark blue cluster of non-neurons at far right:

And Allen’s mouse cortex & hippocampus Smart-seq is similar as well:

Some datasets but not others, then, suggest that Prnp RNA expression is higher in neurons than in non-neuronal cell types, while other datasets show fairly similar expression across cell types. That’s a bit frustrating, but I am comforted by thinking of gene expression on a log scale. In any given tissue, some genes are expressed at 1 TPM and others at 1,000 TPM, and for many genes, one tissue may have 1 TPM while another has 1,000 TPM. So for datasets to agree that Prnp is expressed at roughly the same order of magnitude between different cell types, even if they disagree about which is slightly higher and which is slightly lower, is perhaps not so bad.
PrP protein expression
While it seems there’s little consensus on relative expression of PrP RNA across cell types, it may not matter so much, since what we care about is the protein. Across tissues in the body, which tissues express a gene’s RNA most highly is not a great predictor of which express the protein most highly. One study found that an RNA being low or absent was a good predictor of the protein being low or absent, but that above 32 TPM of RNA expression, there was no correlation between RNA and protein levels [Jiang 2020]. One might hypothesize that the same might be true when comparing across cell types rather than across tissues. Unfortunately, available proteomics methods are simply not as high throughput as RNA expression measurement, and for many analyses, the right types of datasets are only beginning to emerge [Suhre 2021]. The human protein atlas [Thul & Lindskog 2018] evaluated PrP expression across several cell types, but not in CNS tissue — the only neurons examined were in the eye.
A few studies looked at the distribution of PrPC in uninfected brain tissue, but the focus was usually on regional distribution [Taraboulos 1992] or on subcellular distribution — it was found that PrPC in developing brain can be found on neuronal fiber tracts, but in adult brain it is mostly in synapses; in neither cases is it seen on neuronal cell bodies [Sales 1998, Moya 2000]. Those studies did not examine in detail the expression on non-neuronal cell types, however. Immunohistochemistry studies of PrPSc in diseased brain found that PrPSc could indeed be found on astrocytes and microglia [Kovacs 2005], though one couldn’t be certain whether this meant this PrP originated from those cell types, or whether they had simply engulfed plaques or deposits from neurons.
Qualitatively, one can say based on colocalization of PrP with various cell type-specific markers in immunohistochemistry that endogenous PrP in wild-type mice is present on neurons, astrocytes, and oligodendrocytes [Lakkaraju & Sorce 2021] (Figure 2, cropped, wild-type mice, neurons, glia, and oligodendrocytes top to bottom):

But it’s difficult by that method to get to any kind of quantitative estimate of what percent of total brain PrP is contributed by each cell type.
Perhaps the most compelling piece of evidence comes from conditional knockouts. The seminal experiment showing that knockout of PrP after prion infection saves the mice relied on a mouse model with a floxed PrP gene combined with a neuron-specific Cre recombinase expressed under the NFH promoter [Mallucci 2003]. Buried in the supplement is a Western blot showing how much total PrP expression went down after PrP was knocked out at 8 weeks — by eyeballing Figure S3, it dropped by about 80 or 90% [Mallucci 2003]:
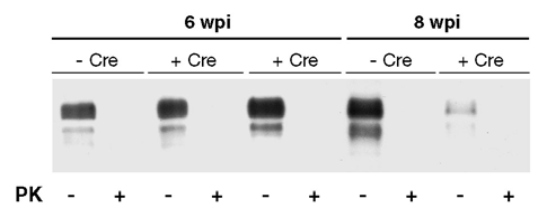
That would suggest that PrP in the brain comes overwhelmingly from neurons, although there are two caveats here. First, I don’t know if there are enough data to be certain that NFH-Cre doesn’t have leaky expression that would have floxed PrP in any other cell types. Second, these mice originally expressed PrP from a transgene based on the cos.Tet vector, not the endogenous locus. That vector has about 24 kb of sequence upstream of the Prnp transcription start site, which obviously is enough to drive robust PrP expression in neurons, but we can’t quite take it for granted that its expression pattern perfectly mimics that of endogenous PrP.
role in disease
We can think of prion disease as involving two processes: prion replication and neurotoxicity. Early evidence suggested that replication stalls out — prion titer plateaus — around the time neurotoxicity begins [Prusiner 1982]. It was therefore suggested that these are two distinct, non-overlapping phases of disease [Sandberg 2011, Sandberg 2014]. As explained here and here, however, the “plateau” may not be perfectly flat, and new evidence shows neuron-specific gene expression changes and rise in plasma neurofilament, suggesting neuronal damage, earlier than previously appreciated [Sorce & Nuvolone 2020, Minikel 2020]. Even if the replication and neurotoxicity phases overlap, though, it’s still useful to think through these two processes separately as we consider each cell type in turn.
neurons
Neurons are what matter in prion disease. They are the only cell type known to suffer any ill effects of prion infection: other cell types, both in the brain (see below) and elsewhere, can replicate prions, but prions don’t kill them, they only kill neurons. Moreover, prion neurotoxicity is cell autonomous. This was shown early on by grafting PrP-expressing brain tissue into PrP knockout mice: the knockout neurons were invincible, even when surrounded by PrPSc [Brandner 1996]. This has replicated more recently in experiments where PrP was re-expressed in specific cell populations of knockout mice using a CAG-CAT system [Lakkaraju & Sorce 2021]. In other words, what matters is the PrPSc produced by neurons, perhaps GPI-anchored to neurons, not PrPSc in the milleu around neurons.
Imagine if a drug could lower PrP expression in neurons to zero, without hitting any other cell types at all. As far as we know today, such a drug would be sufficient to halt prion disease in its tracks. On the other hand, if a drug could only lower neuronal PrP by, say, half, would it still be helpful to also have activity in other cell types? Possibly — read on.
astrocytes
Astrocytes do not suffer from prion toxicity but do replicate prions. There was early confusion on this subject because mice with PrP expressed under the GFAP promoter were reported to develop and die of prion disease after inoculation with prions [Raeber 1997, Jeffrey 2004]. It turns out, though, that GFAP is not perfectly specific to astrocytes, and the transgene construct used in that mouse has some activity in neurons [Marino 2000]. More recent work with the CAG-CAT system produced mice with PrP expression apparently more tightly restricted to astrocytes [Lakkaraju & Sorce 2021]. These mice replicated prions — by 2 years of age they had PrPSc levels similar to terminal-stage wild-type mice. This is in line with the finding that astrocytes can replicate prions in cell culture [Krejciova & Alibhai 2017]. But the astrocyte-specific PrP mice, despite all that PrPSc accumulation, did not develop symptoms nor any neuronal degeneration nor glial activation. In contrast, when PrP was expressed only in neurons, neuronal degeneration and glial activation did occur. Thus, while neurotoxicity is cell autonomous (it requires neuronal PrPSc), glial activation in prion disease is strictly non-autonomous (it, too, requires neuronal, not glial, PrPSc).
These data suggest that if a drug could lower PrP in neurons to zero, you might not need to care about astrocytes at all. But if you can only lower neuronal PrP by half, then it might be nice if the drug also lowered astrocyte PrP, since astrocytes can also replicate prions.
oligodendrocytes
One paper expressing PrP under the myelin basic protein (MBP) promoter found that oligodendrocytes were incapable of replicating prions [Prinz & Montrasio 2004]. Inoculum injected into their brains was cleared just like in PrP knockout mice. (Expression of PrP on oligodendrocytes was reported to actually be helpful against a proteinopathy caused by interstitially deleted PrP [Radovanovic 2005], but we still don’t know whether that proteinopathy in any way models the neurotoxicity of genuine prion disease). I couldn’t find any more recent studies examining the role of oligodendrocytes in prion infection in vivo. In the recent CAG-CAT paper, the researchers also produced mice with PrP expression restricted to oligodendrocytes using the Plp promoter [Lakkaraju & Sorce 2021], but, as far as I can tell they never inoculated them with prions to see what would happen.
microglia
Prion disease has a marked neuroinflammatory response, with high levels of microglial activation. There has therefore been intense interest in determining whether this microglial response to prion infection is harmful or protective. Multiple studies have examined this issue across different experimental paradigms and the conclusion is overwhelmingly that microglia are protective in prion disease. Killing off microglia (with a gancyclovir-CD11b-HSVTK system) accelerated neurotoxicity in prion disease, both in cultured slices and in vivo (50 vs. 57 days median survival time) [Zhu & Herrmann 2016]. IL34 knockout mice, which have fewer microglia, also succumb to prion disease about 10% sooner (162 vs. 176 days for females, 170 vs. 191 days for males) [Zhu & Herrmann 2016]. Treatment with PLX5622, pharmacological inhibitor of microglial receptor CSF-1R, which killed 78-90% of microglia, likewise made mice die of prion disease sooner (~130 vs. 160 days) [Carroll 2018]. A study with mice where an enhancer is deleted to halt microglial development (Csf1rΔFIRE) showed the most dramatic differece of all: about 120 days for microglia-deficient mice vs. 170 for normal mice [Bradford 2021]. Meanwhile, it doesn’t seem to matter much whether the microglia express PrP, as they don’t seem capable of replicating prions. This was studied by irradiating mice and then engrafting bone marrow to introduce exogenous precursor cells to populate the brain with microglia [Priller 2006]. Putting PrP knockout microglia into the wild-type brain didn’t change anything, and putting wild-type microglia into the PrP knockout brain wasn’t sufficient to sustain prion infection [Priller 2006].
conclusions
PrP RNA is expressed to some degree across different brain cell types, though there is little agreement on its relative abundance by cell type. Limited available data suggest that the protein itself comes predominantly from neurons. Neurons both replicate prions and suffer neurotoxicity, and are the one cell type a PrP-lowering drug will really have to reach in order to be effective against prion disease. Astrocytes can replicate prions but their prions don’t seem to contribute to neurotoxicity, so as a second priority it might be beneficial to also knock down PrP on astrocytes. Oligodendrocytes don’t seem to replicate prions and don’t play any obvious role in prion disease. Microglia don’t replicate prions, but their presence (regardless of PrP expression) does play a protective role in prion disease.
What about biomarkers? We will want to measure PrP in cerebrospinal fluid (CSF) to measure the degree to which we’ve lowered it in brain. This will be critical for dose-finding in the clinic and maybe even for a path to provisional approval [Vallabh 2020]. But what we care about most is how much we’ve lowered PrP in neurons specifically, and maybe we also care a little bit about whether we’ve lowered it in astrocytes. Luckily, even if neurons are just half of cells in the brain [von Bartheld 2016], they may contribute disproportionately to PrP protein expression [Mallucci 2003]. If true, then neuronal target engagement will tend to dominate the biomarker readout. Meanwhile, you only really worry about disconnect between the biomarker and the cell type of interest if a drug exhibits cell type-specific activity; a drug active across cell types will tend to have bulk tissue target engagement proportional with neuronal target engagement. For antisense oligonucleotides, at least, available data suggest target engagement across multiple cell types [Jafar-Nejad & Powers 2021], but we need to look at this question in greater detail and the question will arise for future PrP-lowering modalities as well.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.