A cryptic exon to lower PrP
We’ve just posted a pre-print nominating another mechanism by which it might be possible to lower PrP — by use of a “cryptic exon” [Gentile 2023]. In this brief post I’ll unpack the idea in a bit more detail.
motivation
To treat or prevent prion disease, we want to lower PrP. Our first shot-on-goal for lowering PrP is an antisense oligonucleotide — and hopefully soon. Oligonucleotides are amazing therapeutics in terms of their programmability and their specificity. But they also have limitations, such as the rather limited drug distribution to deep brain regions after intrathecal dosing [Jafar-nejad & Powers 2021]. For more than a decade now, Sonia and I have wondered, could there also be a way to lower PrP using a small molecule? What on Earth would the mechanism of action be of such a molecule? We spent fruitless years searching for a small molecule to bind PrP and eventually concluded the task was next-to-impossible [Reidenbach 2020]. What else then?
prospects for drugging splicing
An emerging area is the concept of using small molecules to drug not the protein product, but the RNA, as summed up in a recent review of both the science and the state of play in biotech [Garber 2023]. The supplement kinetin has long been known to correct the splicing defect in ELP1 (formerly known as IKBKAP) that causes familial dysautonomia [Slaugenhaupt 2004, Hims 2007, Axelrod 2011]. In that sense, the concept has been around for years. But many scientists didn’t take seriously the idea that one could design a drug to be specific enough to be safe and effective.
For me personally, my interest was first piqued in 2015 when I saw a lecture by Susanne Swalley about Novartis’s development of a molecule called branaplam [Palacino 2015]. Novartis had run a cleverly designed phenotypic screen to detect molecules that would alter splicing of exon 7 in SMN2 — the same thing that the ASO nusinersen does. Out of 1.4 million compounds they found “very few” hits, suggesting they had zeroed in on a specific mechanism. They found that the hits bound a protein-RNA complex, the U1 snRNP, and they characterized the sequence motif of splice sites affected by the lead compound. It wasn’t perfectly specific — genes other than SMN2 also went up or down — but it was good enough to enter clinical development. Branaplam went on to a Phase 1/2 trial in spinal muscular atrophy, paused for safety reasons, re-started, then got canned for business reasons. Soon, however, Novartis published that they had discovered that branaplam also lowers HTT, the gene involved in Huntington’s disease [Keller 2022]. The mechanism was fascinating: it caused a piece of intronic sequence that looked kind of like an exon to be spliced into the mature mRNA, causing a frameshift. A “poison exon” or a “cryptic exon”, some called it. It was an incredibly enticing proof of concept for how to knock down a gene with a small molecule. This launched branaplam back into clinical development, this time for Huntington’s disease. Development of branaplam for Huntington’s later got stopped for safety reasons, but remember, failure is the norm in drug development, and doesn’t always mean the concept was inviable. By the time branaplam stopped, PTC Therapeutics had a similar molecule for Huntington’s, PTC-518, in the clinic, and Skyhawk had another, SKY-0515, in preclinical development, which just reached Phase 1 recently.
I focused on Huntington’s disease above because the idea of exploiting cryptic exon to lower a target gene was so enticing as a strategy for prion disease. But whether any of the above molecules will succeed is an unknown, and indeed, since tominersen’s Phase 3 failure there is even some doubt about huntingtin lowering as a therapeutic hypothesis. There’s no doubt, far and away the best proof of concept for drugging splicing with a small molecule comes from a different drug: risdiplam. Like branaplam, it was designed to cause exon 7 inclusion in SMN2 for spinal muscular atrophy. Risdiplam showed successful Phase 3 results and reached FDA approval in 2020. This is, to this day, our evidence that splice-modulating small molecules can indeed be specific enough to be safe and effective.
It is probably a combination of the proof of concept from risdiplam, the evidence of different available mechanisms from stories like branaplam, and advances in basic science, that caused biotech capital to flow into this space. As that review mentions, several startups have built platforms around targeting RNA with small molecules, including PTC, Skyhawk, Arrakis, and Ribometrix, and several big pharmas like Novartis either built their own capacity or made deals with those startups. For these companies, success may require a combination of building the right platform — the right phenotypic screens, the right target compound decks, a good structural understanding of the binding event — but also choosing the right targets to go after. It won’t matter how good your molecule is, if it turns out that lowering huntingtin isn’t actually the right idea. And here we have this ideal target, PrP, that we know for sure is essential for disease initiation and progression and where all the evidence suggests no worries about on-target toxicity.
There was just one problem. PRNP has just a single protein-coding exon. How on Earth could tinkering with splicing ever lower PrP?
finding a weakness in the PRNP transcript
One day I saw Sonia hunched over her laptop, spending a weird amount of time poring extremely carefully over an old paper [Lee 1998]. “I think there’s something here”, she said.
The PrP gene in mice, sheep, and most other mammals has 3 exons — 2 that contain only 5’UTR and 1 that contains the coding sequence. Humans and other primates have just the equivalent of the 1st and 3rd exon. The 2nd one is missing. That old paper had observed that the exon 2 sequence actually still existed in the genome, it just wasn’t spliced into the mature mRNA.
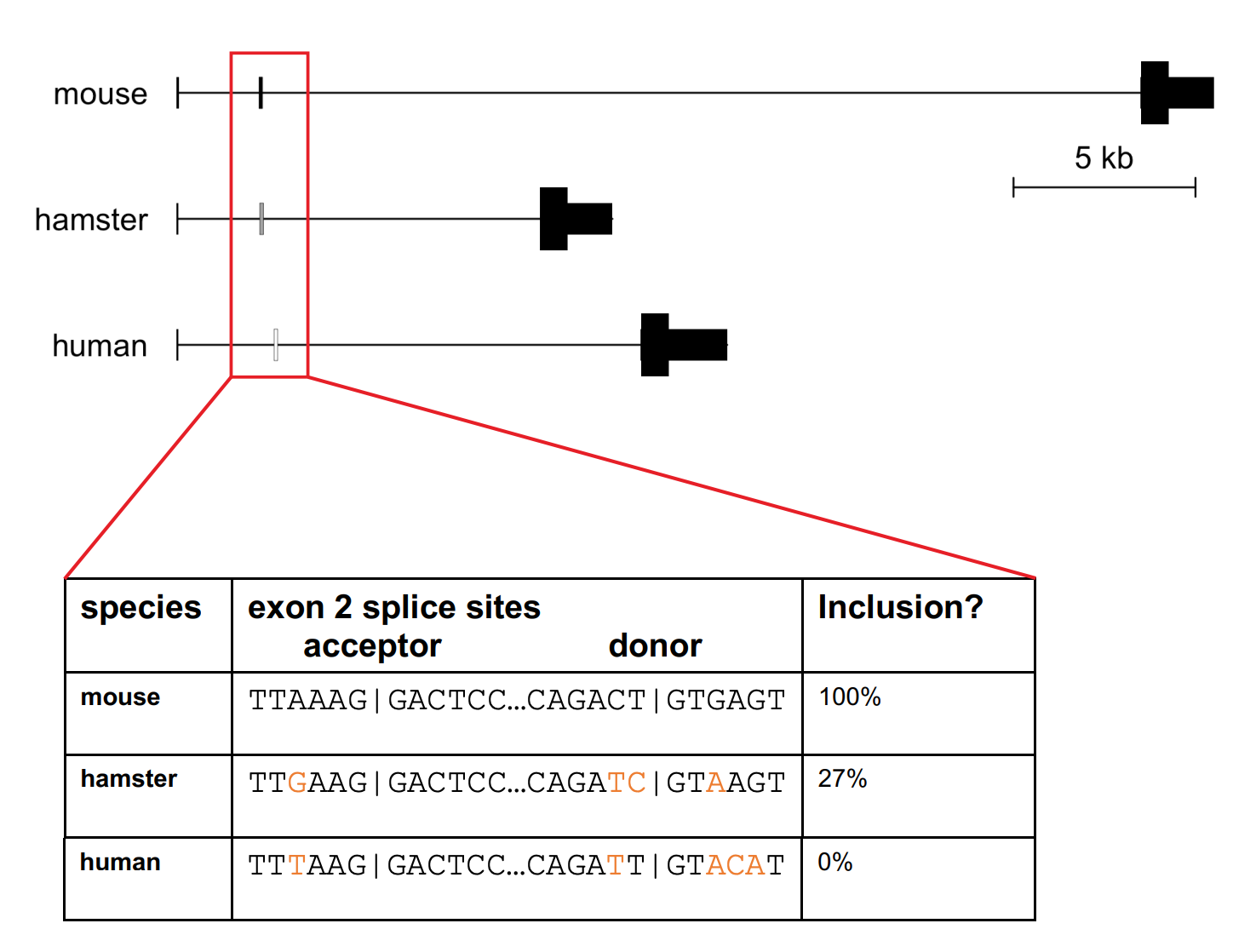
Figure 1B from [Gentile 2023]. The human PRNP gene contains a sequence homologous to rodent exon 2, with essential splice sites intact, but it is never spliced into human PRNP mRNA.
If we could cause it to be spliced in, would that have any functional impact? Sonia had noticed that the human version of exon 2, unlike that of mice, had a start codon, an ATG, in it. There was evidence that having an upstream open reading frame, or uORF — meaning a “decoy” start codon before the “real” start codon of a gene — could reduce the amount of protein translated off of the RNA [Calvo 2009]. Was there a chance that including human PRNP’s exon 2 could lower PrP by this mechanism? I had a friend and collaborator from my human genomics days, Nicky Whiffin, who was an expert in 5’UTRs and had found that genetic variants that create uORFs are selected-against in disease genes [Whiffin 2020]. We called Nicky up and she was happy to advise on the project. From all the analyses we did together, it seemed plausible enough to be worth testing.
Our postdoc Juliana Gentile took up the mantle of figuring out whether exon 2 mattered. It was not trivial to set up a system to study this. If we did CRISPR editing, you would have to subclone, which was both laborious and might result in seeing a lot of clonal differences. We wanted a transfection system where we could study different transfected versions of the gene side by side. But most people do transfection with tiny plasmids containing only the coding sequence, under a blazing strong promoter. Here we needed enough intronic sequence to include “exon 2” plus some context around it, and since the goal was to detect reductions in protein expression, we didn’t want to overexpress to the point of saturation where differences might become undetectable. Juliana settled on a 6.5 kb “minigene” construct — kind of annoyingly large, but still tractable — with some intronic sequence retained and some deleted, and the moderately strong PGK promoter. She codon optimized the coding sequence so that we could distinguish it from endogenous PRNP and designed primers to look at exon 1-2, 2-3, and 1-3 junctions, then designed a series of variants with different mutations in the exon 2 splice sites.
It worked! Strengthening the splice sites around exon 2 caused the exon to be included, and protein expression to go down. This is probably due to the repressive effects of the uORF, but it may be a bit more complicated. Only the variants with multiple nucleotide changes led to appreciable exon 2 inclusion; single point mutations we tested also lowered PrP protein expression (albeit more modestly), but without measurable exon 2 inclusion by qPCR. It could be that nonsense-mediated decay is also at work, or even that some of the mutants caused retention of additional intronic sequence. The best effect was a 78% reduction when we mutated the splice sites to match the most canonical human splice motif. That’s pretty exciting!
what’s next
We’re a highly translational lab and we do a lot of in vivo work — it’s hard for us to stop at just a cell culture result. But after much consideration, we simply couldn’t come up with an elegant way to confirm the effect of exon 2 inclusion in vivo. You would need to make two parallel mouse lines, one with wild-type human sequence — the whole 15 kb of transcript — knocked in, and another with that same knock-in sequence but with the splice sites mutated. That’s a big lift and a long road. And anyway, it wouldn’t address the biggest open question, which is whether splicing of exon 2 can actually be modulated by a small molecule. That’s a question that can only be answered by finding such a small molecule. The splice sites of PRNP exon 2 don’t look like those of ELP1 or SMN2, so there’s no starting point for medicinal chemistry — you’d really have to screen from scratch. Remember that when Novartis found branaplam, they screened 1.4 million compounds and found “very few” hits. The best-designed screen (which that one was) can cut down on false positives and time wasted on follow-up, but still, you need either a very large library or a very focused library of compounds in order to have any hope of finding something with this one very specific mechanism. It’s not something we’re going to achieve at an academic scale, especially given lessons learned from our last effort to find a small molecule [Reidenbach 2020].
Thus, the question becomes whether this proof of concept, together with all the evidence supporting PrP lowering generally, is enough to convince a company to invest in this strategy. The answer could be yes, it could be no, or it could be let’s wait and see how other splice-modulating small molecules play out in the clinic and when the biopharma industry becomes less strapped for cash. There may not be an immediate answer, but we’re in it for the long haul, and to have one more possible way to lower PrP and treat prion disease, we feel it’s worth getting these findings out there and seeing what’s possible.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.