Uniqure reports that AMT-130 slows Huntington's disease
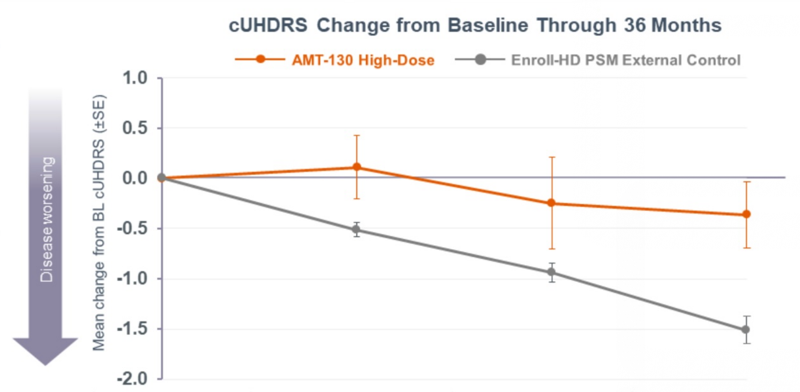
Above: change in cUHDRS, the primary endpoint of Uniqure’s Huntington’s disease trial.
If you’re anyone who spends time thinking about therapies for genetic diseases of the brain, you’ve recently been inundated with the news about AMT-130, the first genetically targeted therapy for Huntington’s disease to report positive results in a pivotal clinical trial. This is big and exciting news in the Huntington’s disease field but, contrary to the hopes of many people who’ve emailed me over the past week, carries no appreciable implications for our work in prion disease. Read on.
background
Huntington’s disease (HD) is caused by a CAG repeat expansion in exon 1 of the HTT gene, encoding the huntingtin protein [Tabrizi 2020]. The length of the CAG repeat is a major determinant of age of onset, for instance 44 CAG repeats typically leads to mid-life onset, while 70 repeats typically causes juvenile onset [Langbehn 2010]. Once disease onsets, it often progresses over a decade or more, with motor, cognitive, and psychiatric symptoms, eventually leading to death. Up to now, there have been no disease-modifying therapies for HD.
HD pathology affects many different parts of the brain, especially as it progresses towards end stage, but is especially intense in the striatum — a deep brain region consisting of the caudate, putamen, and globus pallidus. Several different lines of evidence indicate that the fundamental mechanism of disease pathogenesis is somatic expansion of the CAG repeat. When we say a patient has 44 CAGs, we really mean that they have 44 CAG is the average repeat length in their white blood cells, which is where we are usually sequencing DNA from; it’s probably also the number of CAGs they inherited and had in every cell at birth. But in the affected brain regions, the CAG repeat grows longer over time. In genetic studies, the DNA repair enzymes that control the rate of that somatic expansion have been identified as major determinants of disease onset and progression rates in both humans and mice [Mouro Pinto 2013, GeM-HD 2015, Moss 2017].
The most prominent therapeutic hypothesis in the HD field is that lowering the amount of mutant huntingtin will slow or delay disease [Tabrizi 2020]. Early on, it was discovered that in mice, deleting the mouse equivalent of the HTT gene is lethal [Duyao 1995], but adding back even a mutant copy of the gene will rescue the mice [White 1997]. This meant that even mutant huntingtin can carry out whatever the normal, native function of huntingtin is. This has long been interpreted to mean that the CAG expansion causes disease by a gain-of-function (the mutant gene does some new terrible thing it should never do) as opposed to a loss-of-function (the mutant gene fails to do its original job). This therapeutic hypothesis got a big boost when it was shown that, in mice, using an antisense oligonucleotide (ASO) to lower the amount of huntingtin could slow the progression of disease [Kordasiewicz 2012]. But it was never going to be easy: full deletion of HTT is embryonic lethal [Duyao 1995], and conditionally deleting HTT after mice are born leads to neurodegeneration [Bragg 2024]. There was always going to be a tension between wanting to lower HTT but not lower it too much. One solution would be to lower it allele-specifically, leaving the wild-type allele untouched, but that’s more technically challenging to do, and some approaches would work only for a minority of patients depending on their exact DNA sequence [Southwell 2014]. The first allele-specific drug to reach the clinic (WVE-001) didn’t actually succeed at lowering mutant HTT at all. Ionis Pharmaceuticals adopted the strategy that it was better to just lower all HTT, mutant and wild-type alike, in order to have a more potent and more universal drug. They developed a non-allele-specific ASO called tominersen to lower all HTT — both mutant and wild-type — and licensed it to Roche, who brought it forth to a Phase 3 trial. The outcome was terrible: HD patients treated with tominersen actually did worse than patients on placebo.
Tominersen’s failure led to a lot of questions in the HD field. Why did it fail? Clearly, it wasn’t just ineffective, it was toxic — at least at the combination of dose level, dosing frequency, and patient population studied in that trial (Roche still thinks it might work in some patients). The toxicity might have been due to some off-target protein-binding property of the ASO, and not related to HTT at all. But some scientists I talked to at that time wondered whether it was a case of too little huntingtin — if HTT had been lowered too much, at least in some cells or some brain regions — or if, gasp, HTT lowering wasn’t even the right idea to begin with. Tominersen was designed to target only the full-length HTT transcript, so some scientists pointed to the longstanding theory that a fragment of the HTT mRNA containing only exon 1, where the mutation is, actually drives the disease [Sathasivam 2013], and said that tominersen failed because it didn’t lower exon 1. More recently, a single-cell sequencing study of the striatum in HD patients’ brains gave us a new perspective on somatic CAG expansion [Handsaker 2025]. Every neuron in the striatum has its own unique CAG length which grows randomly over time. Even rather long CAG repeats, such as 80, do not appear intrinsically toxic at the single cell level. Instead, it appears that the CAG repeat has to get extremely long — more than 150 repeats — before neurons start to become sick. The CAG then expands very rapidly until they die by about 500 repeats. At any given instant, most individual neurons are either still below 150 repeats, or are already dead. There are only a minority of neurons right in the sick range. If so, then even if mutant huntingtin is causing a gain-of-function, lowering huntingtin might only help that small minority of currently “sick” neurons, while not actually preventing the not-yet-sick neurons from undergoing further somatic expansion into that sick range. Some scientists took this to mean that the best therapeutic strategy in HD was to prevent somatic expansion — say, by binding or editing the DNA itself at the HTT gene, or by targeting those DNA repair factors.
In the years since all the above developments, until recently, no other pivotal clincial trials in HD had a readout that would answer the question of whether HTT lowering could be beneficial. Then, two weeks ago, we heard a big announcement about AMT-130 from Uniqure.
the concept and the animal data
AMT-130 is an AAV gene therapy developed by Uniqure, a gene therapy company based in Lexington, MA. The therapeutic substance is a viral vector, AAV5, engineered to contain an artificial microRNA (miRNA or miR) that targets and lowers the HTT mRNA. It targets HTT exon 1 and so it lowers both full-length HTT and the exon 1 RNA fragment. AAV5 does not cross the blood-brain barrier (BBB), instead, the vector is injected directly into brain tissue. In neurons that take up the virus, the viral DNA is expected to stick around at least for years, maybe indefinitely, and continue producing that miR that will reduce HTT. According to the company website, the dose is administered “by drilling two to six small holes in the skull”. I could not find any information on the internet as to the exact number of injection sites. 2-6 holes could mean 2-6 injections, or it could be several injections per hole, made at different depths and/or angles. An HDBuzz post from around when the trial launched suggests that it is probably 6 injection sites:
the surgery itself is an 8-10 hour procedure, guided by MRI, involving the insertion of very tiny tubes into six locations deep in the brain.
AMT-130 appears to have been in development for at least a decade. The earliest paper I could find about the strategy [Miniarikova 2016] was submitted in September 2015, which means the experiments were probably underway at least 3 years before then. They were presenting it at HD conferences by 2016 and they got an IND (permission to initiate clinical trials) cleared by FDA in 2019.
Uniqure has published animal data in several species. Using mini-pig model of HD [Baxa 2013], they examined biodistribution, huntingtin lowering, and disease biomarkers [Evers 2018, Valles & Evers 2021]. They also published GLP toxicology studies in monkey and rat [Spronck 2021], though these species are not sequence-matched for miHTT so it wasn’t possible to look at target engagement. In the pig model, the miR matches the HTT target sequence and so they were able to examine target engagement, or lowering of HTT. They reported that mutant huntingtin was lowered to <25% of the original level in striatum. They even saw target engagement in the cortex, perhaps because cortical neurons that innervate the striatum took up the virus, yielding about 50-75% of the original level of mutant huntingtin depending on what part of cortex you looked at [Valles & Evers 2021].
I’ve tried to compile into one table all the information necessary for thinking through how the results from that preclinical pig study might scale to human patients:
| species | total dose | injection sites | brain mass | striatum volume |
|---|---|---|---|---|
| pig | 1.2e13 vg | 4 | 180 g | 1.5 cm3 |
| human | 6e12 vg (low) and 6e13 vg (high) | 6 | 1400 g | 6 cm3 |
Table 1. Comparison of dose level, injection sites, and relevant brain size metrics in preclinical pig model versus humans. Sources: pig and human total brain size from Brain Facts and Figures. Human striatum volume [Yin 2009] and pig striatum volume [Fil 2021] determined by MRI. Dose levels from preclincial study [Valles & Evers 2021]. Dose levels (in viral genomes, vg) from human trial [NCT04120493].
Our human brain size is driven disproportionately by our huge cortex. Non-cortical regions of human brain are larger than other species too, but by a smaller fold change. There’s a 5-fold scaling factor for the total dose from pig to human (high dose) and a 4-fold scaling factor in striatum size. The number of injection sites, however, was only scaled up 1.5-fold. Can just 6 injection sites reach the larger human striatium? This question led me to take a deeper dive how far AAV can diffuse from an intraparenchymal injection site.
AMT-130 is delivered by convection-enhanced delivery (CED), which as far as I can tell is simply a fancy way of saying it’s delivered under slightly high pressure so that the material diffuses further into brain tissue than if ejected slowly at low pressure. There is a ton of literature on CED, but after extensive Google Scholar searches, I found only one paper that was willing to put a number on how far AAV can diffuse from the injection site: “our MR-guided CED infusion techniques show vector spread of up to 10 mm diameter from the injection site for cortical CED” [Yazdan-Shahmorad 2018]. The truth is, though, there is probably no single diameter: vector accumulation is at its most intense right at the injection site, is somewhat diminished a 5 mm radius away, and might be low but non-zero another 5 mm away from that. A neuron that has an axon passing right near the injection site might have taken up virus and end up expressing a viral transgene in its cell body that is centimeters away. Let’s have a look at some data:
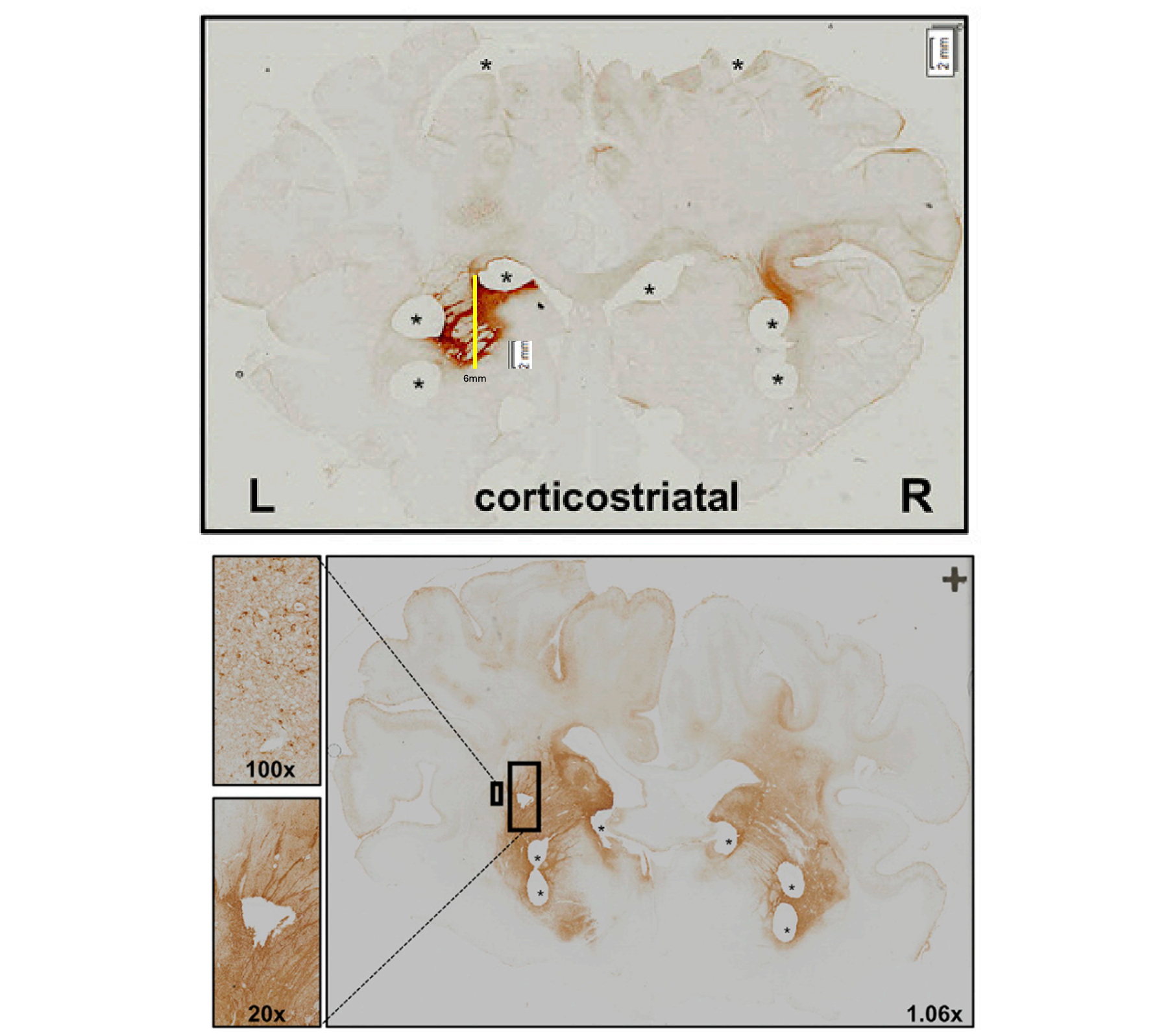
Figure 2L (top) and 4A (bottom) from [Evers 2018]. Expression of a GFP transgene from AAV5 injected into the pig brain at a dose of 1e12 vg (top) or 1e13 vg (bottom).
When I line up a ruler with the scale bar on Figure 2L of [Evers 2018], shown above, and measure what I see as being the transduced area is for a AAV5-GFP transgene in the left putamen (yellow line added by me above), I come up with a diameter of 6 mm, but it’s patchy: some spots within that diameter are totally lacking GFP, while you can find tufts of GFP here and there outside that diameter. The spot isn’t especially round, but as a rough approximation, if the transduced tissue were spherical, its volume would be (4/3)πr3 = 113 mm3, which is 0.1 cubic centimeters. That’s at a 1e12 vg dose. Figure 4A examines the higher 1e13 vg dose, but with no scale bar provided. Eyeballing it, the diameter might be 10 mm, and there are certainly more wisps of GFP-positive cells outside that diameter than at the low dose. A sphere of 10 mm volume works out to 0.5 cubic centimeter volume. If so, then 6 injections of 1e13 vg might reach 3 cm3, or about half the human striatum. Before you jump to speculating whether half is or isn’t enough, remember, this is all just ballparking off of a GFP reporter gene — the data I cited above simply indicate where can one see GFP expression on histological images [Evers 2018]. It’s conceivable that low levels of transduction that aren’t super dark when one stains for GFP, could still be enough to produce some miHTT and some lowering of HTT. Indeed, the data from Figure 3 of [Valles & Evers 2021] suggest an average of at least 10, sometimes as much as 1,000, copies of miHTT per cell in the striatum, with a lot of variability depending on which exact brain punch you look at. That doesn’t mean every cell has them — an average of 10 could be all cells have 10, or half of cells have 20. But it’s consistent with the idea that the injections went a substantial way towards saturating the striatum.
the clinical trial
Uniqure’s trial launched in 2019, and they waited until they had 29 patients treated (17 at the 6e13 high dose and 12 at the 6e12 low dose), each followed for 3 years, before they annnounced the data in a press release. I registered to watch the announcement webcast, and found that the slides contained no copyright claim and I was not required to sign any terms and conditions notice to view them, therefore, I have gone ahead and posted a PDF of the slides for perusal and discussion.
All of the comparisons that Uniqure reports in the press release are based on the N=17 high-dose individuals, compared to a natural history cohort selected from ENROLL-HD. Key points:
- The primary endpoint was cUHDRS, a clinical rating scale that tries to capture everything that goes wrong in HD. (This was also the primary endpoint for tominersen and in many other HD drug trials.) Lower is worse. cUHDRS change was -1.52 in the natural history controls and -0.38 in the high dose patients. That’s a 75% slowing of progression, P = 0.003.
- Each of the various clinical endpoints and measurements (TFC, SDMT, SWRT, TMS) that makes up the cUHDRS moved in the same favorable direction, and some but not all were individually significant.
- CSF NfL, a biomarker of neurodegeneration, was 8% below baseline at the end of 3 years. Although there was no control group for this, Key Opinion Leader Sarah Tabrizi commented in the webcast that typically CSF NfL would rise 35-40% in HD patients in the same time period.
In support of the CSF NfL claim, the below figure shows some natural history data on CSF NfL from patients with manfiest (symptomatic) HD:
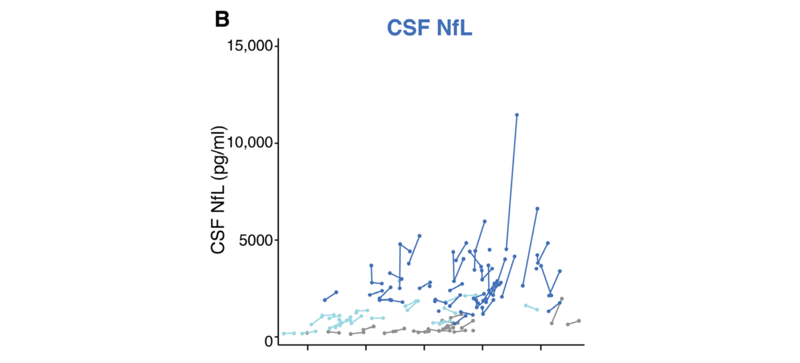
Figure 2B from [Rodrigues & Byrne 2020]. Dark blue points and lines show individual CSF NfL trajectories for manifest HD patients.
Here’s a table comparing the 3-year numerical movements in endpoints between the high dose, low dose, and external controls (separate matched groups for high and low) from ENROLL-HD.
| group | cUHDRS ↓ | TFC ↓ | SDMT ↓ | SWRT ↓ | TMS ↑ |
|---|---|---|---|---|---|
| high dose | -0.38 | -0.36 | -0.44 | 0.88 | 2.01 |
| low dose | -1.65 | -0.33 | -6.44 | -3.67 | 8.64 |
| ENROLL-HD (high match) | -1.52 | -0.88 | -3.73 | -6.98 | 4.88 |
| ENROLL-HD (low match) | -1.72 | -1.04 | -3.35 | -5.20 | 5.61 |
Table 2. Comparison of reported endpoints for high and low dose groups and ENROLL-HD controls. ↓ indicates endpoints for which lower scores indicate worsening of disease, ↑ indicates endpoints for which higher scores indicate worsening of disease.
You can see that on all 5 metrics, the high dose people experienced substantially less disease progression than the ENROLL-HD controls. For low dose people, only TFC looks better than the ENROLL-HD controls, while the other metrics are similar or sometimes worse. Overall there’s no obvious story of efficacy at the low dose.
In all the data presented, the ENROLL-HD natural history cohort are the only controls. Uniqure’s page about the trial states that “10 [participants] did not receive any study treatment (“Imitation Group”)” and the press release also mentions “patients… randomized to… an imitation (sham) procedure (n=10)” and later states that an “additional four control patients crossed over to treatment”. This suggests to me that there remain at least 6 untreated individuals who would be internal controls within the study, but their data are never shown.
As you can appreciate, the evidence of efficacy here depends entirely on the ENROLL-HD controls being well-matched. The following table shown in the slides suggests that the groups are indeed well-matched on all the observable demographic and baseline disease characteristics:
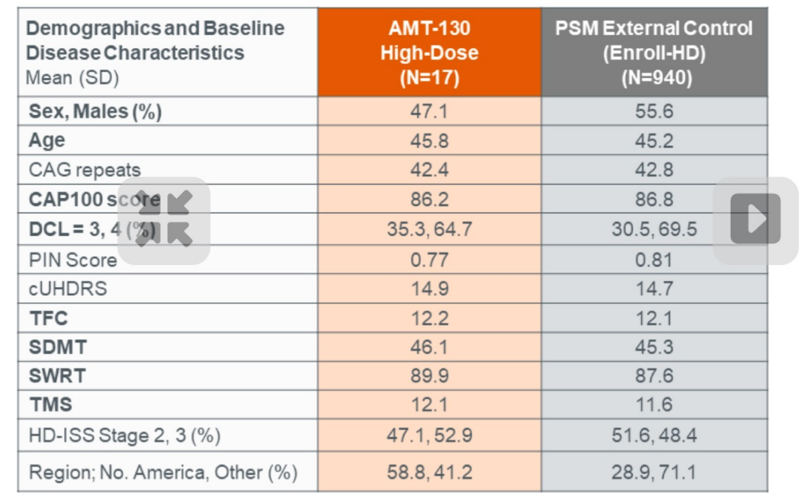
The trial was small while the ENROLL-HD cohort is large, and so each individual in the trial was matched to multiple controls based on various characteristics; as you can see in Table 2 above, the high and low dose groups therefore had slightly different control groups for comparison.
Like most clinical trial press releases, this one is not perfect. Here’s my best attempt at a balanced look at the amount and quality of data presented so far.
Reasons to be skeptical:
- Most of the patients in the Uniqure trial were in open label arms, so there could be some placebo effect, perhaps mediated by belief in the therapy and greater effort to do well on the tests. (This is a common objection raised by FDA scientists to open label trials.)
- Individual data points are not shown in any of the plots; error bars are shown but never defined.
- The data from imitation/sham procedure patients are not shown and their absence is not explained.
- There are no data on target engagement (mutant HTT lowering), which would greatly inform our prior for whether the therapy worked. (Of course, they could have chosen to measure CSF mutant HTT, but since the therapy is only expected to be active in a relatively small brain region, the overall CSF level might well stay constant or increase, even if the therapy is highly potent at decreasing mutant HTT in the brain tissue that it reaches. So it’s reasonable that they excluded it, but still a limitation in our interpretation of the available data.)
Reasons to believe:
- The table in the screenshot above suggests that the trial participants and natural history controls were well-matched on all observable demographic and baseline disease characteristics.
- The high dose patients did a lot better than the low dose patients. If there is any placebo/effort effect, the low dose patients would be equally susceptible to it, and some of the high and low dose patients were in blinded randomized arms.
- Uniqure stated that the natural history comparison statistical analysis plan was pre-specified and agreed upon with FDA in a Pre-IND Type B meeting. FDA is generally very hard-nosed about such comparisons, so their approval of this plan would not have come without significant scrutiny.
- Sarah Tabrizi, world expert on HD natural history and clinical trials and a very rigorous physician-scientist, strongly endorsed her belief in the results as being real. I assume that she knows more details than have been publicly disclosed as to how the ENROLL-HD control matching was done, and has reason to believe it was done well.
- CSF NfL was below baseline at the end of 3 years, which (much as I don’t always believe NfL) is not susceptible to placebo effect or effort effect, and would never be expected within the natural history of HD. If you look at the natural history data for CSF NfL above, it’s noisy and sure, occasionally someone’s value may go down here and there, but it’s quite unlikely by chance that you’d select any 17 patients such that the average of NfL goes down over 3 years.
Overall, then, I find more and stronger reasons to believe than not to believe. In particular, Sarah Tabrizi and Ed Wild, two HD scientists who I deeply respect, have publicly stated their belief that the result is real. The HDBuzz post about the trial readout takes a very optimistic tone with a hint of caution:
This is the very first time any drug for HD has shown statistically significant slowing of disease progression on clinical measures accepted and in alignment with the FDA. That makes the results very encouraging (and the HDBuzz editorial team reach for a tissue as we happy cry our way through writing this article).
Even with all of this good news, it’s important to be cautious. Firstly, the number of people treated in this trial is still very small. All of the statistics reported in this update relate to data from less than 30 participants, only a portion of whom received the high dose of the drug that seems to show benefit. Further, many of the comparisons made to show how well this drug is working were against an external control group, not participants within the same trial. Although carefully matched, this kind of comparison is not as strong as a classic placebo-controlled study. There are also some pieces of the puzzle which are missing. This drug is designed to lower huntingtin levels, but there is no report in this update that the drug is working to do that… We also didn’t learn anything about how this drug might impact different regions of the brain structure from imaging analyses like MRI.
My plan, then, until we see more data in a publication, is to hold just a bit of space in my head for doubt, but proceed on the assumption that probably this is real — AMT-130 may well be the first disease-modifying therapy for HD.
what comes next in HD
Uniqure says they plan to file a BLA (biologics license application) with FDA in Q1 2026, meaning that next year, there could already be an approved, disease-modifying drug available to HD patients. That’s absolutely huge news for people suffering from HD today.
This also validates the therapeutic hypothesis of HTT lowering. Indeed, it may give us some information about the nature of the dose-response relationship between HTT lowering and disease modification. While we don’t have human target engagement data, the lowering of HTT in the human brain from 6 injections into 6 cm3 of striatum in a 1400 g brain couldn’t possibly look better than the pig data from 4 injections into 1.5 cm3 of striatum in 180 g brain. Thus, the data from [Valles & Evers 2021] probably put an upper bound on how good the data look: at best, reduction of mutant HTT to 25% of its original level in striatum, and perhaps to 75%, maybe as little as 50% in some cortical regions. The Uniqure data, then, suggest that this is enough to achieve 75% slowing of disease progression. Alternatively, consider this: when Roche designed the tominersen trial, they thought they needed hundreds of patients to power it. Uniqure’s data would suggest that HD responds so robustly to HTT lowering that just 17 patients plus a natural history control arm is enough to get a highly significant result.
How can that be, and what does that mean? Well, remember that tominersen actually made patients in that trial worse, so it’s not solely a matter of being ineffective. Three hypotheses as to why AMT-130 did so much better might be:
- AMT-130 focused on striatum, which is a relatively harder-to-reach brain region for an intrathecal ASO like tominersen.
- AMT-130 targets exon 1, which tominersen does not.
- Maybe tominersen was actually great in terms of on-target effects but these were just outweighed by some toxicity.
Depending on the answer, the Uniqure announcement is really good news for some or all of the other HTT-lowering therapies in the pipeline, of which there are several — see for instance the HDBuzz posts on SKY-0515, PTC-518 (+company slide deck), ALN-HTT02 (+ company slide deck), WVE-003. These are all different therapeutic shots on goal, which will have varying levels of target engagement in striatum, and some but not all of which will target exon 1.
If the AMT-130 data are indeed as good as they sound then, it seems likely we will hear other good news from HTT-lowering trials over the coming years. And while the striatum is especially hard-hit in HD, many brain regions are affected, so it’s possible that some of these drug candidates, which may achieve broader distribution across the brain, may do even better.
implications for prion disease
Despite how exciting all this is for HD patients, I don’t see any obvious implications for prion disease.
The main reason is that prion disease is a whole brain disease. There is no single small structure in the brain that we could target in order to have an outsize effect on the disease process. We need to lower PrP everywhere. Yes, there are example where a particular subtype of prion disease is particularly intense in one brain region, but by end stage — which comes very soon because this is such a rapid disease — profound pathology is everywhere. If each intraparenchymal injection can reach a half cubic centimeter of brain tissue, then no, I don’t think anyone’s doing 2800 injections directly into the brain of prion disease patients.
The only way we’re going to achieve a 1-time AAV-vectored therapy to lower PrP in the whole brain is if we can cross the BBB. Last year we saw a big breakthrough with the report of the first AAV designed to be administered systemically — into the blood — and reach the whole brain. Then last month, we saw a huge setback, with the death of the first patient ever dosed with a BBB-crossing engineered AAV, shortly after dosing. That’s both a tragedy for that family and disease community, and undoubtedly a setback for the whole AAV engineering field. Capsida, the company that developed that AAV and was running that clinical trial, has so far been secretive and not disclosed much about what happened. We still do not know what receptor their AAV targets, what the patient’s cause of death was, or what dose level of AAV they administered. Those are key variables. Until we know more, it would be premature to abandon our investment in BBB-crossing AAVs. It could be that other BBB-crossing AAVs, such as that one I blogged about last year (which has still not reached a first human dose), may be safer, for example either because they use different receptors, or because they can be dosed lower. The death in the Capsida trial is not a reason to pivot to direct injections of AAV into the brain for prion disease — not given the size of the brain that we need to reach.
All that being said, the Uniqure results are a good reminder of a couple of important lessons about clinical trials.
First, natural history data, describing how patients decline over time in the absence of treatment, are incredibly powerful for enabling good clinical trials. We owe a huge thanks to everyone in the prion disease community who has participated in such natural history studies, for instance, Brian Appleby’s TAPCJD study. The Uniqure story also illustrates that sharing of such data is absolutely essential. Uniqure could never have done this trial by simply pointing to a curve in a figure in a paper and saying “look, our patients are different than those patients”. The primary endpoint of this trial depended on access to individual patient data in order to match each patient in the trial to a comparator group of natural history patients who were similar on key dimensions such as age, CAG length, and disease stage. In order for natural history data to be useful, the individual patient-level data must be available to the companies and investigators running the trials and with the regulators tasked with evaluating their efficacy.
Second, drug development takes time, and many shots on goal. HTT lowering has been a therapeutic hypothesis since the 1990s, and the first mouse study showing that HTT lowering could be beneficial was published in 2012. It’s only now, 13 years later and after weathering the failure of tominersen, that we finally get clinical validation that HTT lowering works. And there is surely still a lot more to learn about HTT lowering, as we follow treated patients for longer and as we see the results of additional HTT-lowering drugs. As we all eagerly anticipate the readout of the first PrP-lowering drug trial, perhaps by end of this year, we can both hope for the best possible outcome, and also know that the arc of history is long.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.