Fatal familial insomnia as a sleep disorder
 Read with caution! This post was written during early stages of trying to understand a complex scientific problem, and we didn't get everything right. The original author no longer endorses the content of this post. It is being left online for historical reasons, but read at your own risk. |
Fatal familial insomnia is a prion disease, more broadly a protein-folding disease and a neurodegenerative disease, and those are the angles from which it is most often studied. But it is clearly also a sleep disorder. I have never been clear on why FFI affects sleep: is it just because it kills neurons in the thalamus, where sleep/wake is regulated? Or do FFI prions directly interfere in sleep pathways? So I decided to dig up some reviews on sleep science and see what the world knows about sleep at the molecular level.
I started with a review by Cirelli 2009 and was surprised to learn, in the second sentence, that FFI is considered a big deal in sleep science. When Medori 1992 (ft) linked FFI to the D178N mutation, PRNP became the first gene known to be involved in sleep. (Fatal familial insomnia had already been characterized – and named – by Lugaresi 1986, but the causal gene wasn’t yet known then). And PrP’s connection to sleep is not just through FFI either. Tobler 1996 and Tobler 1997 (ft) showed that PrP knockout mice have subtly altered circadian rhythms and sleep EEGs, a point I’ll return to later in more detail. And Landholt 2006 has also noted sleep phenotypes in familial CJD.
As I got deeper into Cirelli 2009, I realized I needed to start from a more basic first step: what is sleep, physiologically and molecularly speaking? Cirelli & Tononi 2008, in an article provocatively entitled “Is Sleep Essential?”, define sleep as follows:
Sleep is a reversible condition of reduced responsiveness usually associated with immobility. The decreased ability to react to stimuli distinguishes sleep from quiet wakefulness, while its reversibility distinguishes sleep from coma.
This definition is intentionally general to allow it to encompass species very different from us. In humans, sleep is usually characterized based on EEG waveforms (Cirelli 2009 Fig 1b):

“Spindles” are the really dense, short waves seen in stage N2 above. The loss of them is characteristic of fatal familial insomnia. They are clearer on this Wikipedia graphic:
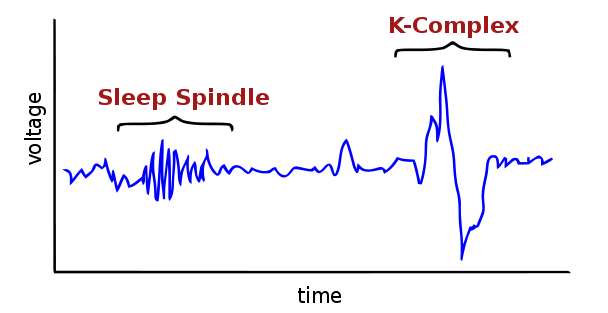
Spindles are characteristic of Stage 2 sleep; each stage of sleep in humans has its own characteristic EEG appearance as well as behavioral features. Sleep quantity and quality are often measured in EEG terms; if you ever participate in a sleep study they’ll probably measure your brain activity with an EEG in addition to your heart rate, eye movements, blood pressure and other things.
Researchers have quantified these features in other mammals and birds as well [Cirelli 2009] and they seem to at least partially fit the human conception of sleep in terms of brain activity. For other species it is somewhat more logistically difficult to record electrical activity in the brain, though incredibly, people have done it, even in fruit flies [Nitz 2002]. To get at a broader swath of the animal kingdom, researchers now define sleep in terms of (as described above) reduced activity and reduced responsiveness to stimulation.
Many scientists have assumed, for decades, that all animals sleep by this definition. And sleeping is a dangerous business. As Sehgal & Mignot 2011 put it:
As a state that seemingly freezes all productive activity and puts animals in danger of being caught by predators, sleep must serve an important purpose because it has survived many years of evolution.
If we believe that all animals sleep, despite the high cost to doing so, then there must be some really fundamental biological role that sleep plays which is conserved across all animal species. If so, scientists still don’t know what that role is. Therefore Cirelli & Tononi 2008 open by challenging the idea that sleep is essential, proposing that perhaps the reason we haven’t found its core function yet is because it has none. They toy with this idea and then propose that if the null hypothesis is actually true – sleep has no core essential function across species – then we should be able to find at least one animal that never sleeps. Or at least, an animal that doesn’t need extra recovery sleep following sleep deprivation, or an animal for whom sleep deprivation does not have serious consequences.
Yet Cirelli & Tononi 2008 argue that in fact there is no convincing example of an animal that doesn’t sleep. Every animal that’s been studied intensively does have periods of reduced activity and reduced responsiveness, and does get extra ‘rebound’ sleep after a period of researcher-induced sleep deprivation. They also all do have serious health problems (eventually death) if chronically sleep deprived, with the possible exception of the pigeon. But the difficulty in studying that last point is that it is almost impossible to completely deprive animals of sleep. Humans, properly motivated, can stay awake for several days – it’s terrible for their health, but it’s possible – whereas no matter how hard researchers try, they can’t seem to keep rodents from sleeping at least a few minutes here and there.
Which brings us to one of the probably-universal facets of sleep in the animal kingdom, which is that animals all seem to experience ‘sleep pressure’. There are two distinct genetic systems that regulate sleep pressure: circadian clock and sleep homeostasis. The circadian clock is a set of genes that enforce a 24-hour cycle, while the sleep homeostasis system enforces the total quantity of sleep. These are two different things and can disagree with each other – for instance, if jetlagged, you might feel sleep pressure from your circadian clock even when you’ve had adequate sleep, or might not feel sleepy even when sleep deprived, if your circadian clock thinks it’s daytime where you just came from.
The circadian clock genes are expressed predominantly in the suprachiasmatic nucleus, part of the hypothalamus. In humans (but not in all animals) they not only enforce a 24-hour cycle but also help to consolidate sleep into a single large block. The same core mechanisms of 24-hour cycling appear conserved at least between humans and fruit flies if not among an even larger swath of the animal kingdom. In terms of human genes, the basic mechanism of molecular timekeeping (as I’ve come to understand it from these reviews and Wikipedia) is that CLOCK and ARNTL trigger transcription of the period and cryptochrome genes PER1, PER2, PER3, CRY1 and CRY2 which, once translated in the cytoplasm, gradually form complexes and migrate back to the nucleus where they shut off their own transcription. Remember, this is the circadian clock, not the sleep homeostasis system: sleep deprivation doesn’t shift the phase of this 24-hour cycle.
None of these reviews addressed the fact that people are capable of adjusting to jet lag (or to working night shifts). Clearly, it’s possible to reprogram the circadian clock in the long term.
The circadian clock is the best-understood piece of this whole puzzle. According to Sehgal & Mignot 2011, we don’t know quite as much about the molecular / genetic basis of sleep homeostasis. However we do know how to observe it: sleep homeostasis has a really robust EEG signature. EEG is discussed in terms of power spectrum: the amplitude of each of the different wavelengths that the overall waveform can be decomposed into. Interestingly, different people have different EEG power spectra (which can be measured during wake or sleep) and the differences appear to be 70-90% heritable [Cirelli 2009]. Despite these differences, everyone has certain signatures of sleep: for instance, the very deepest sleep (slow wave sleep or SWS) is characterized by high amplitude of the ‘delta‘ wavelength (~1 to ~4 Hz). The amplitude of delta waves (dubbed “delta power”) when you first reach deep sleep is tightly correlated with how much sleep pressure was experienced before finally sleeping. Sleep deprivation means more powerful delta waves once you finally sleep, and naps mean less powerful delta waves when you finally bed down for the night. Because delta power seems to correlate with sleep need rather than time of day, it is believed to reflect sleep homeostasis rather than the circadian clock, and can be used as a proxy for homeostasis-induceed sleep pressure [Maret 2007].
What sleep pressure consists of at a molecular level is not as clear. But we do know some genes and molecules that appear to be involved in sleep and in sleep pressure, which Sehgal & Mignot 2011 group into a few categories.
First, the ion channels. Neurons in the cortex and thalamus are hyperpolarized during sleep (in mammals), meaning the amount of stimulus required to make a neuron fire an action potential is higher than during wake. So there is a very simple ion concentration reason that explains part of why animals are less responsive during sleep. Something similar is probably going on in fruit flies, since mutations in their voltage-gated potassium channels make them sleep much less (2-4h/day instead of 8-10), though still on a 24-hour cycle (the circadian clock isn’t disrupted). The fly gene is Shaker, corresponding most closely to the KVβ1 subunits responsible for IA (A-type K+ current) in mammals. Mutations in potassium channels as well as calcium channels have some sleep phenotypes in mice, though much less notable than in fruit flies. That’s probably because mammals have a lot more genetic redundancy. Apparently Drosophila‘s lack of redundancy, and thus dramatic phenotypes from single gene knockout, are one major reason why people study Drosophila.
The period genes are a key part of the circadian clock as discussed above. PER3, at least, also appears to be involved in sleep pressure. The gene contains a 54bp repeat, of which some human chromosomes have 4 copies and some have 5 copies. A couple of (admittedly small sample size) human studies suggest that 5/5 homozygotes experience harsher sleep pressure after sleep deprivation [Viola 2007, Groeger 2007].
Then there are neurotransmitters. In mammals, adenosine and GABA are pro-sleep, while histamine, dopamine, acetylcholine, and norepinephrine are all anti-sleep. All of this makes sense with what you know about drugs. Caffeine is an adenosine antagonist. Valium and other benzodiazepines are GABA partial agonists. Benadryl, an anti-histamine, makes you drowsy. Nicotine is an agonist for nicotinic acetylcholine receptors. Cocaine stimulates by inhibiting reuptake of dopamine and norepinephrine (and serotonin, whose role in sleep is more complicated). Those are just the small molecule neurotransmitters; then there are also neuropeptides (short peptide neurotransmitters). Of these, orexin (aka hypocretin) is known to have an important pro-sleep role; narcoleptics don’t have enough of it, for reasons we don’t fully understand. There are actually two different orexin peptides – orexin-A (33 amino acids) and orexin-B (28 amino acids), both processed from the protein product of the HRCT gene. Hrct knockout mice exhibit narcolepsy, but curiously, and we don’t know of any mutations in the human HRCT gene associated with narcolepsy – instead, it looks like orexin deficiency in narcoleptics might be due to autoimmune attacks on the orexin peptides themselves [reviewed in Nishino 2007].
There are a wide range of other genes whose transcription level correlates with sleep/wake cycle – perhaps 2,032 transcripts in all [Maret 2007]. Genes involved in energy metabolism are upregulated during wake [Cirelli 2009] – no surprises there. Some of the other findings are pretty interesting though. For instance, Maret discusses four mouse genes with roles in neuronal recovery from “glutamate-induced neuronal hyperactivity” which get upregulated in response to sleep deprivation, . These genes (Homer1a, Ptgs2, Jph3, and Nptx2) appear to be helping the neurons restore calcium homeostasis after repeated stimulus. Otherwise neurons can die from being flooded with too much calcium after being stimulated over and over, a phenonmenon called excitotoxicity. Another intriguing angle is that heat shock proteins and chaperones are up-regulated during waking and that sleep deprivation may trigger the unfolded protein response [Cirelli 2009]. Indeed, heat shock proteins appear to help Drosophila survive sleep deprivation [Shaw 2002]. It is tempting to tell a good story about all this: maybe waking produces molecular waste (such as misfolded proteins, and excess calcium in the cytosol) faster than neurons can cope with it. Maybe sleep, then, is a necessary break for neurons to get their house in order so they’re ready to face another day.
But getting sold on one story would be quite premature at this point: there are a lot of different theories, all of which look pretty decent. Another angle is that genes involved in synaptic potentiation (increase in sensitivity to stimulus) are upregulated in wake, and genes involved in synaptic consolidation and depression are upregulated in sleep. So maybe learning is a two-phase process: make new connections by day, prune and organize by night. As Cirelli puts it:
One idea is that sleep consolidates synapses activated by learning during the previous waking period, perhaps through mechanisms that require cortical spindle activity and the slow waves of NREM sleep. Another possibility is that, since wakefulness is associated with a diffuse potentiation of synaptic circuits that results in a net increase in synaptic weight, sleep produces a generalized depression of synapses. This downscaling would benefit the brain because it decreases the energetic cost of synaptic activity, eliminates weak and ineffective synapses, reduces cellular stress and increases signal to noise ratios.
Whatever the mechanism, there seems to be little doubt sleep is important for learning, and that people learn better when they’re caught up on sleep.
A number of other pathways of genes appear to be upregulated during sleep: membrane trafficking and maintenance, myelination, cholesterol synthesis, possibly even protein synthesis generally. It’s less fun to tell stories about these things, but as scientists we have to remember we don’t know whether one, some, all, or none of the observations discussed above constitute a core function of sleep.
While reading in these reviews about all the genes that either follow sleep-wake cycles, show sleep phenotypes in animal models, or have been implicated in human sleep disorders, I noticed the mention of RARB - retionic acid receptor beta, a master transcription regulator which binds retinoic acid, a derivative of Vitamin A, and is known to be crucial in embryogenesis. Apparently it is also important for sleep: Maret 2005 examined the “DBA/2J” (D2 for short) inbred strain of mice, which are severely deficient in delta power in their sleep EEGs, and was able to link this deficiency to a mutation in the 5′UTR of the mouse Rarb1 gene, leading to a decrease in Rarb1 transcription.
The reason that caught my eye is that Grizenkova 2010, studying a large number of outbred mice, found that polymorphisms in the Rarb region were associated with prion incubation time, and Mead 2009, in a genome-wide association study for vCJD risk, found a SNP near RARB to be the #1 most-nearly-significant hit. Neither of those studies was quite a home run – neither study could definitively link the association to the RARB gene, just the general region, the GWAS hit wasn’t quite significant after multiple testing correction and the mouse study only looked at one strain of prions (RML prions). And even if RARB is associated with prion disease risk / incubation time, there’s no evidence yet to suggest that it’s part of the mechanism by which PRNP affects sleep.
Indeed, from what I read in these reviews, it doesn’t seem that we know much of anything about prion protein’s role in sleep. I dug a bit deeper into Tobler 1996 and Tobler 1997 (ft) to understand the sleep phenotypes of the PrP knockout mice.
It is well-established that when mice are put in complete and permanent darkness, their circadian clocks gradually shorten to a ~23 hour day instead of 24 hours. Tobler 1996 basically showed that PrP knockout mice stay pretty steady at 24 hours no matter how long they stay in the dark. To show that this was definitely PrP-related, Tobler also tested PrP knockout mice with a PrP transgene introduced back into the genome, and indeed, those mice were more like the unmodified controls. Very interesting – I would love to know if the PrP knockout mice therefore can’t adapt to jet lag either.
That paper also hinted at a few other sleep phenotypes of PrP knockout, but those are more thoroughly explored in Tobler 1997. To summarize, Tobler found that PrP knockout mice don’t sleep any less than controls, but they do have more interrupted sleep. When they sleep, they have more ‘brief waking episodes’ – waking up for 4 – 16 seconds and then going back to sleep – and also their NREM sleep is in a greater number of shorter installments interspersed among the REM sleep. PrP knockout mice didn’t reach as strong of delta power during NREM sleep, but they did fall into NREM sleep more quickly, reaching their own peak delta power sooner than controls, and they also respond to sleep deprivation with more dramatically enhanced NREM sleep than the controls do.
Tobler suggests a slightly complicated explanation for all this:
the null mice have a low sleep pressure, which leads to (1) more frequent interruptions of sleep, and (2) low amounts of SWA. This interpretation is supported by the lower amount of SWA reached within NREM sleep episodes in the null mice. As a consequence, the propensity for SWA can increase more during SDEP in these mice. This notion is consistent with the findings in humans, in which habitual long sleepers displayed a larger increase in SWA after SDEP than habitual short sleepers (Aeschbach et al., 1996). It was concluded that the short sleepers live at a higher SWA pressure and, therefore, that SDEP cannot enhance SWA to the same extent as in the long sleepers. Because TST in the mice was not different, it is possible that cellular mechanisms responsible for the increase of SWA are affected by the lack of PrP. The absence of PrP, therefore, seems to affect sleep via two mechanisms that may be interrelated: (1) a decrease in sleep pressure, and (2) a diminished capacity to sustain NREM sleep. In addition, the faster increase of SWA at the transitions from waking to NREM sleep in the null mice may reflect a difference in the underlying cellular mechanisms leading to this transition because of the lack of PrP.
So this is kind of hard to wrap your head around – somehow, PrP is needed for baseline sleep pressure but is not involved in sleep pressure in response to sleep deprivation. (If PrP were a crucial part of all sleep pressure, then the knockout mice wouldn’t have had any increase in delta power after sleep deprivation – let alone a larger increase than controls.) To me, it is hard to reconcile this explanation with the fact that, according to Tobler’s earlier paper, PrP knockout mice actually have a more reliable circadian clock than controls – you’d think that therefore they must experience intense circadian sleep pressure.
To me it is clear that PrP influences sleep but not clear what its exact role is. A number of studies since Tobler’s finding have explored this connection further and as far as I can tell, there have not been any major breakthroughs. Sanchez-Alvarez 2007 replicated most of Tobler’s findings and found that PrP expression specifically in neurons and not in glia mattered for the sleep phenotypes. Plazzi 2002 suggested that PRNP codon 129 genotype (MV vs. MM) affects EEG power spectra in pre-symptomatic FFI carriers, but Pedrazzoli 2002 failed to find any strong relationship between sleep phenotypes and codon 129 in the general population.
It is also unclear whether PrP’s native function in sleep has anything to do with fatal familial insomnia. It does not appear that the FFI genotype itself causes any sleep abnormalities: Ferrillo 2001 found no evidence that healthy asymptomatic carriers of FFI have abnormal or impaired sleep. Sleep does not seem to be disturbed in FFI until other things get disturbed as well. Cortelli 2006 found subtle sleep EEG abnormalities 13 months before onset in one patient – the same time as glucose hypometabolism in the thalamus was visible via FDG-PET. (The patient’s EEG had been normal at a clinical visit 21 months before onset). Are the EEG changes and glucose hypometabolism due to FFI prions impairing neuronal function, or because neurons had already started dying outright? Cortelli is straightforward about not knowing whether the abnormalities visible 13 months before onset are due to some neuronal loss (but not enough for clinical symptoms) or due to neuronal impairment but not yet death of neurons.
The most thorough discussion of sleep and fatal familial insomnia appears to be Schenkein & Montagna 2006 Part I and Part II. Part I describes the exact nature of the insomnia experienced in FFI:
As described by Montagna,[34] sleep in FFI is characterized by an early and progressive reduction in sleep spindles and K-complexes, a reduction in total sleep time, and disruption of the cyclical organization of sleep; SWS is lost first, then REM disengages from its circadian cycle and intrudes into the waking state. Lugaresi and colleagues[47] explain that circadian rhythms involving melatonin gradually decrease and shift in phase, and finally disappear. The rhythmicity of somatotropin (GH) shows a similar reduction or total loss in tandem with the loss of deep sleep. Only prolactin rhythmicity remains unaltered.
The authors note that insomnia can cause memory problems, thalamic hypometabolism, weight loss, and so on. In short, some (though not all) of the other symptoms of FFI could theoretically be explained as the result of insomnia. But it’s not clear whether that’s actually the case. The authors report:
Despite its suggestive name, the insomnia of FFI may not be an early or essential symptom of the disorder. Among a series of German patients, sleep disturbances were mild and often recognized only in retrospect after detailed questioning of the family or reinvestigation of the hospital records. Similar observations have been reported in other international populations.
So in many cases, the other symptoms of FFI – cognitive decline, confusion, ataxia, weight loss, etc. – were so pronounced that family members barely noticed that the patient was not sleeping. On the other hand, Part II presents the case of a man who began suffering insomnia and only later developed memory problems. After being genotyped for D178N and testing positive for FFI, he:
was 10 months into his illness (stage I). He resolved to make the most of his life before the onset of stage IV. DF purchased a motor home and embarked on a solo tour of the United States. He typically drove great distances, but only after a refreshing sleep; he would stay in rest stops for several days until again renewed by sleep. Before embarking, he required himself to recall many numbers, including his date of birth, social security number, etc, and drove only if he remembered all of these.
That’s a far cry from most FFI patients, who barely live 10 months from onset. For this patient it sounds as though insomnia was the first and foremost symptom, with other issues arising later and being ameliorated on the occasions when he was able to get a good night’s sleep. So it would seem that the prominence of insomnia among the many symptoms of FFI varies greatly between patients.
Another question is why circadian rhythms are lost in FFI even though the hypothalamus (as opposed to the thalamus), where circadian clock genes are active, experiences little to no neuronal loss. The authors offer two possible (not mutually exclusive) explanations: the hypothalamus innervates the thalamus, so its signals to the rest of the brain may be lost due to damage to the thalamus, and/or, the hypothalamus may be impaired by misfolded PrP even though neurons don’t die. In support of that possibility, proteinase K-resistant PrP is found in the hypothalamus in FFI. Of course, PK-resistant PrP is probably not a good proxy for toxicity, and the authors note that in FFI it is found in many brain structures (such as the brain stem) where neurons are not lost. (In fact, the authors state that the total amount of PK-resistant PrP shows little correlation with neuronal damage across brain regions, or with clinical severity across patients.)
While it’s clear that prion replication is really what is destroying the brain, the authors discuss the possibility that if insomnia contributes to patient decline in FFI, then treating the insomnia (if possible) might prolong life or alleviate symptoms. Indeed, this has been attempted using gamma-hydroxybutyrate, a CNS neurotransmitter that induces sleep. GHB, as it is known, was (according to WebMD) available as a supplement in the U.S. until it was banned in 1990, then got FDA approval as a prescription drug for some symptoms of narcolepsy in 2002; it is also abused as a street drug. One FFI patient [described in detail in Reder 1995], fourteen months into the disease course (i.e. quite advanced) was given GHB every night for 2 weeks and was able to get about 3 hours of slow-wave sleep per night – whereas most FFI patients get none. After the GHB treatment began, he became much more lucid – “Whereas before receiving this medication, the patient was unresponsive to his environment, when he awoke from his drug-induced sleep, he was alert, attentive, and responsive to questions” [Schenkein & Montagna 2006 Part I]. After 2 weeks the doctors stopped the GHB treatments and the patient abruptly died; it wasn’t clear if withdrawl from GHB was a problem or if the patient was simply at the end of the road either way, after 14 months of disease. In any event, the fact that this patient became more lucid after treatment is taken to suggest that at least some of the cognitive symptoms of FFI are downstream of insomnia.
The case report in Part II is quite a story. The FFI patient described therein lived with the disease for 26 months and apparently was completely lucid at least some of the time, all the way up to the end. He had a PhD in alternative medicine and aggressively self-medicated with over-the-counter supplements as well as drugs he was able to get from his treatment team at Massachusetts General Hospital. At various points throughout the illness, he treated himself with huge doses of vitamins, anesthesia, sleeping pills, stimulants (which made him more alert during waking hours and also made him sleep better once they wore off), daily walks, electroconvulsive therapy (ECT), sensory deprivation chambers, and meditation. These things were used for varying amounts of time and in different combinations but the patient reported that each helped him to sleep for at least a while, and that his other symptoms were lessened after a good night’s sleep.
Curiously, GHB didn’t work for him. The effect of ECT was mixed: it helped him to sleep, but gave him severe confusion and amnesia, which is common for ECT in general and also concordant with at least one other report of ECT in prion disease.
For 129MM FFI sufferers like this patient, the mean disease course is 12 months – by living 26 months, he was several standard deviations above the mean. This, and the fact that he was so much more lucid after sleeping, does suggest that at least some of his self-treatments were beneficial. On the other hand, the fact that he was mentally together enough to launch all these efforts even when diagnosed 10 months into the illness (a point at which many patients would already be catatonic) suggests his case was already exceptional. Overall, the FFI case reports seem to paint a picture of a broad spectrum of presentations of FFI: some, like this man, experience insomnia first and foremost and other symptoms secondarily, while for others (such as the German families mentioned above) the insomnia is barely noticed.
But regardless of how big a role insomnia plays in the patient’s decline in fatal familial insomnia, it would be very valuable to know why FFI prions cause insomnia. At present we have no idea why different PrP mutations cause prion diseases localized in different parts of the brain. The fact that PrP has been implicated to have a role in sleep invites speculation that a toxic gain of function related to its sleep role might be involved in FFI. And even if that speculation proves unfounded, understanding the molecular basis of PrP’s involvement in sleep would give us fresh hints to PrP’s native function and possibly the pathogenesis of prion diseases.