Genetics 10: 'Genetic screens by example: virulence factor gene regulation'
These are my notes from lecture 10 of Harvard’s Genetics 201 course, delivered by Tom Bernhardt on September 29, 2014.
Last time we discussed allelic replacement as a means for gene targeting in yeast. Today we’ll also discuss recombineering as one alternative approach.
Recombineering
Here we’ll consider the example of creating knockouts.
In Step 1, you clone a kanamycin resistance (KanR) cassette with primers homologous to the chromosomal locus you wish to target, into a plasmid that also contains a repts cassette (see last time), and an array of three λ recombination genes (exo, beta and gamma, all under the Para promoter), which greatly increase the frequency of homologous recombination. Although no live phage is present, the use of these three λ genes leads to this technique sometimes being called “lambda recombineering”.
In Step 2, you grow your bacterial strain of interest at 30°C in LB plus arabinose. You can make your cells “electro competent” (able to survive electroporation) by spinning them down at a low temperature. Electroporate them to introduce your PCR product from step 1 into the cytosol. Plate them on LB with kanamycin at 37°C and then raise to 42°C. At the high temperature the plasmid cannot replicate, so you end up with only the bacteria that have undergone recombination.
Pathogenesis
Some factors that determine the ability of bacteria to infect multicellular eukaryotic hosts are:
- Invasins - secreted proteins that break down host defenses
- Toxins - killing host cells or poisoning eukaryotic proteins whose job is to scavenge nutrients that bacteria need
- Secretion systems - see below.
Type III secretion systems
Type III secretion systems or T3SS, as introduced in BBS 230 week 01 and week 04, are a mechanism evolved from flagella in gram-negative bacteria used for injecting proteins into host cells.
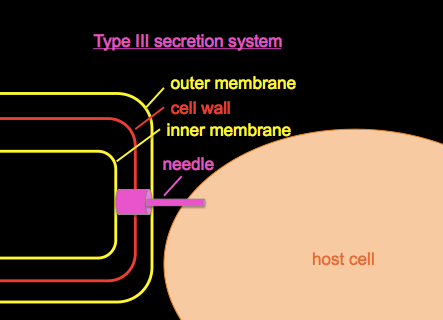
We’ll discuss Pseudomonas aeruginosa, a bacterial pathogen common in hospital-acquired infections and which is often resistant to many antibiotics. It secretes a toxin called ExoU. If you wanted to study it, you could begin by growing a culture of Pseudomonas in a flask of normal LB, centrifuge to obtain a cell pellet, and Western blot with an anti-ExoU antibody to roughly quantify the amount of ExoU present. If you do this you’ll find some ExoU in the cell pellet and none in the supernatant, i.e. a small amount of ExoU is produced but retained in the bacterial cytosol. If you grow your culuture in a flask with EGTA, a Ca2+ chelator, the low calcium environment mimics conditions in the host and appears to have some signaling effect on the bacteria, so if you Western blot again, you’ll find that ExoU production has vastly increased, and that ExoU is now found in both the cell pellet and the supernatant, meaning that some is being secreted.
The ExoU gene is located in an array of genes all involved in the T3SS. Upstream of this array is a promoter which we’ll call PtypeIII, and you want to study the regulation of this promoter, as you don’t yet know whether the stimulatory effect of calcium depletion on ExoU is at the level of transcription. You could perform qPCR on ExoU and/or other members of the gene array to determine whether transcription increases upon calcium depletion. Suppose the answer is that transcription does increase upon calcium depletion. Now you want to figure out the mechanism by which reduced Ca2+ affects translation. To do this, it might help to put a reporter gene under the promoter of interest so that you can monitor the promoter’s activity without doing qPCRs. You decide to use the classic reporter gene, LacZ, so you clone it under the control of the -100 to +10 positions of the PtypeIII promoter. Now you can just measure βgalactosidase activity in the bacteria, and let’s say one flask of culture produces 25 units of activity in normal LB and 1000 units of activity in LB + EGTA. Alternately you could plate them on LB with Xgal, and the low activity (plain LB) bacteria will be white and the high activity (LB + EGTA) cells will be blue.
With this system in hand, you can perform genetic studies to understand the regulation of the PtypeIII promoter. Mutagenize your cells, grow them in plain LB (without EGTA so non-inducing), plate them on LB + Xgal, and you’ll find that most of the colonies are white. Rarely, you’ll get a blue mutant with a constitutively active PtypeIII promoter. The blue colonies must have mutations in proteins that regulate the promoter. These could be, for instance, transcription factors which bind the PtypeIII promoter.
Transposons
Transposons are mobile genetic elements flanked by inverted repeat sequences, and typically encode a transposase and a drug resistance gene. The transposase protein binds to the inverted repeats and cuts them, excising the piece of DNA between them. The transposase then catalyzes the insertion of this DNA into another place in the genome. The choice of insertion site is fairly random.
Transposons can be delivered between cells via:
- Conjugation, which are a natural system
- Transposomes, which are a system of human invention used for mutagenesis.
In a typical transposome protocol, you PCR-amplify a “mini transposon” containing only the inverted repeats and a kanamycin resistance cassette. The mini transposon does not contain the transposase gene, and thus it will only integrate once and won’t keep moving around thereafter, which is a useful property for laboratory studies. You combine the mini transposon DNA with purified transposase, and the protein-DNA complex remains stable for a long time if frozen at -20°C in glycerol devoid of Mg2+ ions. This complex is called the “transposome”. Upon electroporation into cells, the complex becomes activated due to the presence of physiological Mg2+ in the cells.
You can prep some electro competent cells, electroporate them with a transposome, and plate them on LB with kanamycin to select for cells that received a transposon insertion. At the right dilution, most cells will have exactly one transposon insertion. E. coli have ~4,000 genes spread over about 400Mb, so you usually want to create a library of ~100,000 transposon insertions so that you have an insertion on average every 40bp, which gives you an excellent chance of hitting almost every gene several times. Tom endorses the transposome kits sold by Epicentre.
When studying your transposon mutants, you will want to map the location of your transposon insertions by using PCR (more on this next time). You’ll also want to confirm that the transposon is causal for the phenotype of any mutants you’ve identified. You can accomplish this by transduction (i.e. using phages to move pieces of the genome from one strain of bacteria to another, as introduced last time. This lets you recombine the transposon mutation into a fresh reporter strain to see if the phenotype is reproduced. You could also undertake some other confirmatory tests, for example by adding the gene back in to do a rescue, or making a targeted deletion of that gene - but these are hard to interpret without knowing whether the transposon mutation represents a loss-of-function or a gain-of-function.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.