A cautionary tale of ascertainment bias in genetic disease age of onset
I am proud to announce today the publication of my first scientific paper in The American Journal of Human Genetics [Minikel 2014, full text, source code here], and coverage in Nature News.

The backstory is this.
Last April, I saw a new paper come out in PLOS One discussing age of death in genetic prion disease [Pocchiari 2013]. It looked at families with Creutzfeldt-Jakob disease (CJD) caused by the PRNP E200K mutation and found that children died of the disease, on average, 12 years younger than their affected parent had. The authors argued that genetic prion disease must exhibit anticipation — decreasing age of onset in successive generations.
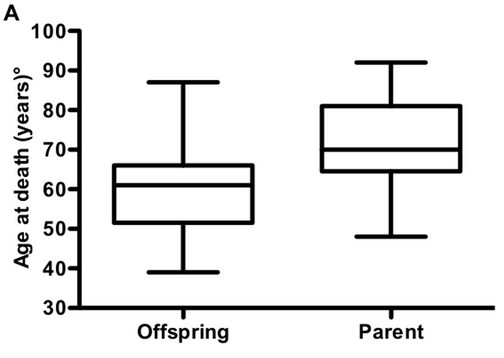
Above: Figure 2A from [Pocchiari 2013], arguing that children die younger than their parents with the E200K mutation.
I found this paper disconcerting on a couple of fronts. First, my wife has the PRNP D178N mutation, which she inherited from her mother who died of genetic prion disease at age 52. The authors weren’t saying anything about the D178N mutation, but it stood to reason that different dominant disease mutations in the same gene might well behave the same - so did this imply that my wife’s life expectancy was in fact 40, and not in her 50s as we had assumed? Secondly, what on earth could be the mechanism for such an acceleration of the disease? As far as we know, prions have only ever been transmitted between people through ritualistic cannibalism, blood transfusions, contaminated surgical instruments and cadaveric transplants or supplements [Gajdusek 1977, Brown 1992, Hewitt 2006] — never through normal day-to-day casual contact. Did the idea of anticipation suggest that maybe people caring for their ailing parents were becoming infected with prions, accelerating their own age of onset? It was a chilling prospect.
Though disturbed, I was also skeptical. I was working as a computational staff scientist in a Huntington’s disease lab at the time, so I knew a bit about how families are ascertained for these sorts of genetic studies, and the types of bias that can arise.
In particular, I knew that most of the research centers that study genetic disease were established relatively recently, and have only been able to collect data for a couple of decades - a tiny window of history. The PRNP E200K mutation was first reported in 1989 [Goldgaber 1989], and in fact, most countries didn’t establish surveillance centers that kept track of prion disease until several years later. That leaves us, at most, 25 years of data collection up through 2013. Consider the two families diagrammed below.
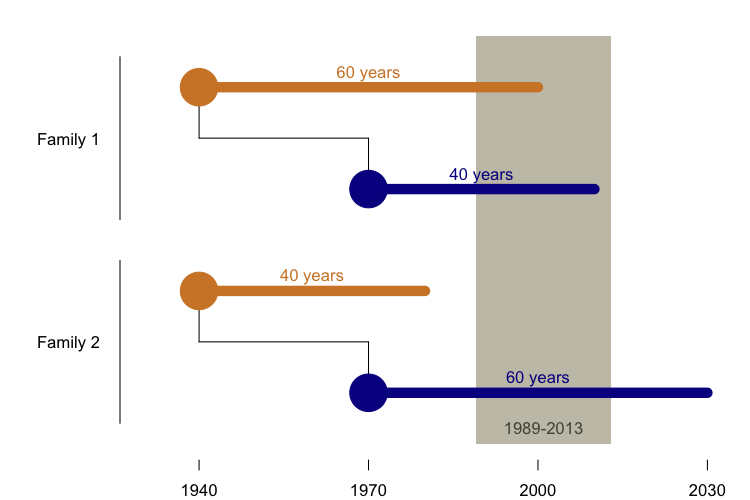
In both families, the parent is born in 1940 and the child is born in 1970, and both individuals have the E200K mutation. In Family 1, the parent dies at age 60 and the child dies at age 40. In Family 2, the parent dies at age 40 and the child dies at age 60.
In Family 1, the deaths occur in the years 2000 and 2010. By 2000, the mad cow epidemic had hit the U.K., people knew what prions were, and CJD was a “notifiable” disease in most developed countries, with all cases required to be reported to national authorities. There is a pretty good chance that the parent and child in Family 1 would both be diagnosed correctly, that at least the child (by virtue of having a family history) would undergo PRNP sequencing, and that the pair would be recorded in some researcher’s spreadsheet as a family with the E200K mutation.
In Family 2, the parent dies in 1980, when neurologists’ tools for diagnosing Creutzfeldt-Jakob disease were crude, the PRNP gene hadn’t been discovered yet, and precious few researchers bothered to keep track of who died of this rare disease. The child will die in 2030, but is still alive and well today, and probably has no idea that she carries a lethal mutation.
Under a null hypothesis of no anticipation, we would expect families like Family 1 and Family 2 to be equally abundant in the world, but when we look at the data that we’re actually able to collect, we’ll overwhelmingly see families that look more like Family 1. In fact, for these hypothetical years of birth of 1940 and 1970, we can only ascertain pairs where the parent lives healthily to at least 49 years old (thus having onset after 1989), and the child doesn’t make it to 43 (thus having onset by 2013). If we then do a t test to compare age of onset in parents and their children, we could very easily fool ourselves into thinking the disease exhibits anticipation.
Could this sort of ascertainment bias could be enough to explain Pocchiari’s results? I opened up a text editor and an R terminal and got cracking. Within a few minutes I had written a simulation to generate 100,000 pairs where the parent’s age of onset and the child’s age of onset were drawn from the same distribution — 64±10 years — but their years of birth were separated by 28±6 years, and you could only ascertain people who got sick between 1989 and 2013.
# simulation
set.seed(1)
parent_yob = runif(n=100000,min=1700,max=2000) # generate parents born from 1700 through 2000
child_yob = parent_yob + rnorm(n=100000,m=28,s=6) # child born when parent is 28+-6 years old
parent_onset_age = round(rnorm(n=100000, m=64, s=10)) # parent's age of onset is 64+-10
child_onset_age = round(rnorm(n=100000, m=64, s=10)) # child's age of onset is 64+-10
parent_onset_year = parent_yob + parent_onset_age # figure out what year parent has onset
child_onset_year = child_yob + child_onset_age # figure out what year child has onset
# if we can magically ascertain _everyone_ regardless of year of onset, is there anticipation?
t.test(parent_onset_age,child_onset_age,paired=TRUE,alternative='two.sided')
# no difference in age of onset
# what if we can only ascertain pairs where both get sick in 1989-2013?
parent_ascertainable = parent_onset_year >= 1989 & parent_onset_year <= 2013
child_ascertainable = child_onset_year >= 1989 & child_onset_year <= 2013
pair_ascertainable = parent_ascertainable & child_ascertainable
t.test(parent_onset_age[pair_ascertainable],child_onset_age[pair_ascertainable],paired=TRUE,alternative='two.sided')
# highly significant 17-year difference in age of onset
Sure enough, the 1989 - 2013 year of onset cutoffs alone were enough to cause 17 years of “anticipation” in a paired t test. If this still doesn’t seem intuitive, consider the following plot, which illustrates another artifact of ascertaining only people with onset between 1989 and 2013:
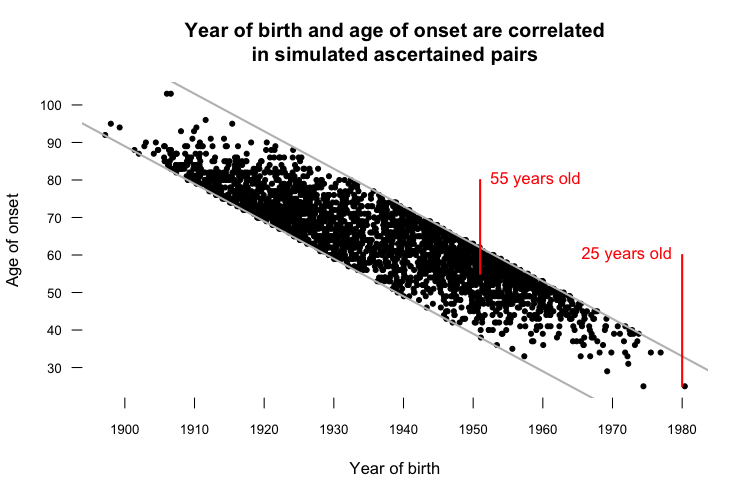
The top diagonal gray line represents onset in 2013, and the bottom diagonal gray line represents onset in 1989. In the simulation, every ascertained data point has to fall between these two lines. That means that the average age of onset observed for people born in the year my parents were born - 1951 - is 55 years old, while the only person born in the 1980s, when I was born, had onset at age 25 (as indicated in red). It also means that year of birth and age of onset are correlated, among the ascertained pairs.
Now consider this: a parent’s year of birth is correlated with a child’s year of birth - for example, a parent born in 1920 probably has a child born in the 1940s or 50s, whereas a parent born in 1950 probably has a child born in the 1970s or 1980s. Because parent and child years of birth are correlated with each other, and year of birth is correlated with age of onset, it turns out to be the case that parent and child ages of onset are correlated with each other.
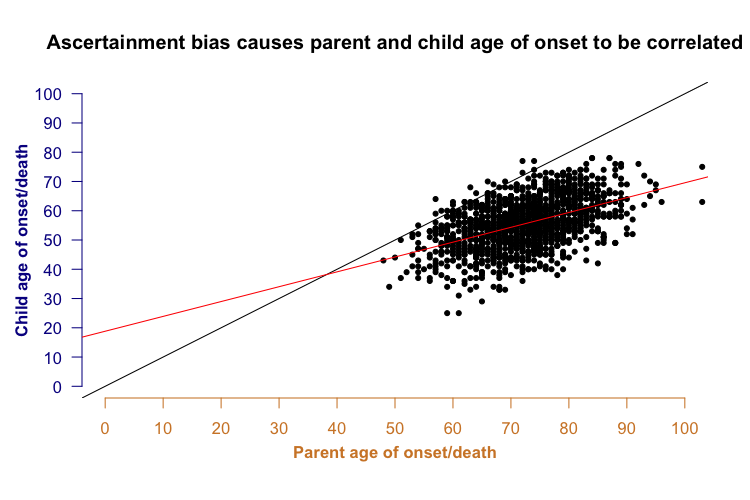
In the plot above, not only do most of the points lie below the black y=x line (indicating anticipation - younger onset in child than parent), but there is also a clear correlation between the parent and child onset (red best fit line). This is a problem because parent-offspring regression has traditionally been considered a way of assessing a trait’s heritability. For many genetic diseases, it has been suggested that in addition to the specific genetic mutation causing the disease, other genetic factors also control the disease’s age of onset. Often, one argument for such “heritability” is that parent and child age of onset are correlated - but in the above plot you see quite a strong correlation even though when I generated the data points for the simulation (see R code above), the parent and child ages of onset were independent. In other words, ascertainment bias alone could be enough to make it look like a disease has a heritable age of onset, even if it doesn’t.
With these simulation results, things were off to an interesting start - but real datasets are far more complicated than those few lines of code reflect. So I spent the rest of the afternoon adding details to my simulation, and soon I ended up spending my next several weekends on it. I varied the size of the ascertainment window, asking how much it would help if instead of 1989, you could ascertain everyone back to 1950 or even 1880. I loaded in the Social Security Administration’s actuarial life tables and took into account the fact that the life expectancy distribution affects what ages of onset we can observe: some people who have the mutation will still die of heart attacks, cancer and accidents before prion disease can get them. I ran repeated simulations with a more realistic N of 100 to see how often I would get significant anticipation. I implemented the same stratification tests that Pocchiari had used to try to rule out ascertainment bias as a problem in his data, and found that his stratifications did not eliminate false positives in my simulation.
As I went, I also started reading the scientific literature on anticipation. There are 70 years of work, going all the way back to [Penrose 1948], showing the ways in which ascertainment bias can create a false signal of anticipation in genetic disease. For many years, Penrose had the world convinced that all reports of anticipation were just biased nonsense. That lasted until the 1990s, when new genetics tools allowed the discovery that some diseases, like myotonic dystrophy and Huntington’s disease, were caused by unstable trinucleotide repeat expansions (think CAGCAGCAG → CAGCAGCAGCAGCAGCAGCAGCAGCAG) that sometimes really did get larger in successive generations, and with increased size, brought earlier disease onset. That opened up a whole new era of inquiry, with a series of papers providing simulations and new statistical tests for analyzing anticipation [for instance: Hodge & Wickramaratne 1995, Heiman 1996, Vieland & Huang 1998, Rabinowitz 1999, Tsai 2005, Boonstra 2010].
None of the literature I found addressed the way in which real datasets are often a messy mix of different ways of ascertaining people. First you have the directly ascertained individuals, people like my mother-in-law who present with the illness and are subsequently revealed to have a causal genetic mutation. Then once you’ve found that first patient in the family (often called the index case), you try to collect a family history, and so sometimes you get retrospectively ascertained individuals - say, a grandparent or an uncle or someone who may well have died undiagnosed but, with the benefit of hindsight, almost certainly shared the causal mutation with the index patient. And then you have the prospectively followed individuals - people like my wife who opt in for predictive genetic testing after their parent dies, find out that they have inherited the mutation, and then keep in touch with the clinician or research center, sending an email every year or two to say “hey, by the way, I’m still alive”.
Many studies of genetic prion disease, including Pocchiari’s, have such a mix, and so next I asked whether the inclusion of retrospectively and/or prospectively ascertained cases would prevent a false signal of anticipation. I simulated the ability to ascertain parents who died before 1989 by collecting the family histories of their children who died later, and the ability to ascertain children who wouldn’t die until after 2013 by doing genetic testing and then treating them as “censored” in a survival analysis at their age in 2013. What I found was that either retrospective or prospective phenotyping could entirely fix the problem - but only if ascertainment was absolutely exhaustive. If you couldn’t always get a full family history, or you couldn’t always follow up on the children who inherited the mutation, then you were still liable to see a false signal of anticipation.
Those are both real problems. Family histories aren’t always collected. Sometimes the clinician doesn’t get around to it, sometimes the family doesn’t want to talk about it. Even when histories are collected, I hypothesized that there is a significant recall bias based on how long ago people have died. People don’t forget their own parents’ deaths, but the index patient isn’t the one giving a family history, because by the time they are diagnosed, they have severe dementia. The family history comes instead from the patient’s spouse or children, whose recall may be less than perfect. My father’s parents both died when I was a child, and today I could not tell you the year of death, age at death or cause of death for either of them. My mother’s parents are still alive today, and when they do pass away, you can bet I’ll remember. Therefore there may be a bias where the longer ago someone died, the less likely a clinician or researcher is to be able to collect data on them.
Meanwhile, prospective follow-up on people who’ve inherited the mutation isn’t perfect either. In a report on a U.K. cohort published earlier this year, less than 25% of people at risk for genetic prion disease chose to get tested like my wife did [Owen 2014]. Therefore, even if one tries to take asymptomatic carriers into account with a survival analysis or similar, it is unlikely to wholly fix the false anticipation problem.
Once I’d taken the simulation as far as I could, I considered blogging my analysis here on CureFFI.org. I imagined a post presenting these simulation results and arguing that Pocchiari’s results might well be false. But a couple of things stopped me. First, though everything I’d seen in the simulation only made me more suspicious that ascertainment bias was behind Pocchiari’s result, I wanted to be sure I was right before I went public with the story, and that meant I had to have a look at some real data. Second, I realized that when I did tell the world, I wanted to make sure the message had an impact beyond the readership of my blog.
The first time Sonia and I met with a genetic counselor was in late 2011, shortly after learning that her mom had the D178N mutation and before Sonia had gotten her own blood drawn for genetic testing. The counselor told us that the disease was dominant, meaning Sonia was 50/50 to have inherited the mutation, and that it was highly penetrant, so that if she had inherited the mutation then she was almost certain to get the disease eventually. (Sonia and I both worked in consulting at the time and had barely retained a high school-level understanding of biology.) We asked a lot of questions about age of onset, and the counselor said there was a wide range of ages of onset for this disease, but at least there was no anticipation. That was the first time I ever heard the term anticipation. After the counselor explained what it was, we asked, “ok, but you’re saying that isn’t something we need to worry about.” She reassured us not to worry, saying “I double checked.”
In that conversation, it was a good thing Sonia had the D178N mutation and not the E200K mutation, because if our genetic counselor had double-checked whether there was anticipation in E200K prion disease, she wouldn’t have been able to offer us any such assurances based on what she saw in the literature. Fifteen years ago, there had already been one report claiming E200K anticipation, published in Neurology [Rosenmann 1999]. That study was from a different research group and studied a different cohort consisting of Israeli E200K patients, but the results nearly identical to those Pocchiari published more recently, showing a 7 to 12 year younger age of onset in parents than children.
Even more worrisome, I learned of a presentation claiming anticipation from yet a third research group based on yet a third E200K cohort, this one in Slovakia. These results weren’t published, but the lead researcher had presented them at the 2012 CJD Foundation conference. Once a year, the CJD Foundation brings together a couple hundred of people personally affected by prion disease for a four-day conference in Washington, D.C. They organize a family support group and opportunities to meet with experts and ask questions, and they also put on one full day of scientific talks intended for a patient audience. A large fraction of the audience at this conference consists of asymptomatic people who have the E200K mutation. In 2012, that audience heard this presentation arguing that age of onset is getting 12 years younger in each successive generation.
With all this in mind, I decided that if I was going to refute the notion of E200K anticipation, I wanted to publish it in a venue where the genetic counselors of tomorrow were sure to find it. And I wanted it to be definitive, so that no one would feel the need to “teach the controversy” and tell their patients that some studies have found anticipation and others hadn’t.
I started calling up all the prion scientists I knew who ran clinical studies or surveillance centers, showing them my simulation results and asking whether they would be willing to collaborate on sharing data on E200K families so that we could see if this “anticipation” was real or not. I’d had many fruitful conversations about age of onset with Simon Mead at the MRC Prion Unit in London, and he had encouraged me to think I would get a warm reception if I reached out to people about data sharing. He was right. Michael Geschwind at UCSF told me he had been present at the CJD Foundation conference in 2012, and that the people in the audience who had the E200K mutation themselves were (he always puts things mildly) “upset,” and that he would like to get to the bottom of this issue. Inga Zerr of Germany’s surveillance center and Steven J. Collins of Australia’s CJD surveillance both came on board as well, saying that if this anticipation business was indeed wrong, then we had to set the record straight.
My collaborators set about to gather up their E200K data, and spent hours on Skype and over email walking through the interpretation of the data with me, categorizing cases according to how they were ascertained and helping me understand the different possible sources of bias in their data. After several months we wound up with what I believe is the largest dataset to date on this mutation: 217 E200K individuals.
The real data look strikingly similar to what I saw in my simulation. Even though all of the four centers contributing data do try to collect family history to some degree, 92% of the years of disease onset in the dataset still fell between 1989 and 2013. Year of birth and age of onset were strongly correlated, again just as in the simulation. We had prospective follow-up data on a number of asymptomatic individuals with the mutation, but overall, less than a quarter of people who were at risk had been tested.
Based on these signs of ascertainment bias, we expected that if we simply applied a paired t test as Pocchiari had done we would see “anticipation” in our data too. But based on the simulation results, we also hypothesized that the amount of “anticipation” we saw would depend on how strictly we allowed our ascertainment to be limited by the years the research centers had been in operation. Sure enough, if we considered only the patients who were seen “directly” at the various centers (no family history and no prospective follow-up), we saw a 28-year paired difference in age of onset. If we loosened the inclusion criteria to allow pairs where one individual was ascertained “directly” and the other was obtained through family history, that figure dropped to 15 years. When we also allowed pairs where data on both individuals were obtained through family history, it dropped to 7 years. The more strictly we limited ourselves to the 1989-2013 ascertainment window, the more anticipation we saw.
Performing a survival analysis with the asymptomatic individuals included didn’t help very much, probably because we had relatively few asymptomatic individuals. Our E200K pedigrees were full of at-risk children and grandchildren of people who had died of prion disease, but we knew the genotypes for less than a quarter of these people. However, if we performed a survival analysis on only the UCSF and MRC Prion Unit cohorts, which had strong family history inclusion as well as the highest numbers of asymptomatic individuals, the “anticipation” dropped to less than 1 year. Even in these cohorts, ascertainment was far from perfect, but these results were enough to suggest that in the limit where ascertainment was as thorough as possible, the “anticipation” would disappear altogether.
I also had the idea that if the “anticipation” in these pedigrees was simply due to ascertainment bias, it shouldn’t be limited to E200K individuals. Some of our pedigrees contained extensive family history data with births and deaths of multiple generations of people, including those who had died of cancer or heart attacks or in wars. Indeed, in some cases where the E200K mutation came from, say, the father’s side, we even had this sort of extensive family history on the mother’s side of the family, which was entirely unaffected. In other words, we had (as Reviewer 1 later encouraged us to call it) a negative control: people who we knew didn’t die of prion disease, but whose ascertainment was still subject to the forces of bias that shaped our whole dataset. Sure enough, when we looked only at the people in our pedigrees who didn’t die of prion disease, they had anticipation too - children died 14 to 35 years younger than their parents, depending on how exactly we defined the inclusion criteria for the analysis.
When all these lines of evidence were put together, it formed a compelling story: there is no evidence for anticipation in E200K prion disease. The reported “anticipation” is solely an artifact of ascertainment bias. That’s a relief to me, and I expect it will be an even bigger relief to people with the E200K mutation, a few of whom have contacted me over email or Facebook in the last year to ask whether I thought the reported “anticipation” was real or not.
Along the way, we also uncovered some issues that should prove to be of broader relevance to people studying adult-onset genetic diseases. Age of onset is a dangerous phenotype. It’s one where a person’s phenotype dramatically affects their chances of being ascertained for a study, and this means we need to exercise a lot of caution when estimating age of onset, testing for anticipation, or determining whether age of onset is heritable. To my knowledge, our study is the first to demonstrate that the dangers of age of onset are not readily mitigated just by making an effort to collect family history or track asymptomatic individuals, and it is also the first to use deaths of unaffected family members as a “negative control.” While some of the dangers of the age of onset phenotype have been studied for nearly 70 years, erroneous claims of anticipation continue to be made. The holy grail might be to develop a statistical test for anticipation that reduces false positives almost to zero without losing any statistical power to detect true anticipation — we didn’t achieve that, and personally, I doubt it can be done. Instead, we provided a common sense walkthrough of the ways that ascertainment bias reveals itself in age of onset data, and I hope this will help other researchers to identify such bias in their own data, and exercise appropriate caution.
As a final point of reflection, it is a pleasure to finally write that blog post I originally thought about writing a year and a half ago. While I love blogging, the experience of turning this into a real study, and not just a one-off simulation, has been immensely rewarding. The analysis in this paper ended up being the job talk that got me hired at the MacArthur Lab in January 2014, and that gave me access to loads of helpful advice from my mentors at MGH - Daniel MacArthur, Mark Daly, Ben Neale, and Mike Talkowski. My collaborators’ critiques of my simulation greatly improved it, and the AJHG editors and Reviewer #1 put an enormously helpful effort into getting us to be more statistically rigorous, more thorough, and to explain our convoluted thought process more clearly. I owe a debt of gratitude to all of these folks. And as one last note, I also owe a debt to all the families whose data are represented here. Having been through it with Sonia, I know that participating in research is a non-trivial effort, but your participation is what allows us to do studies like this one. Thank you.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.