Protein folding 02: Alpha helices and coiled coils
These are my notes from week 2 of MIT course 7.88j: Protein Folding and Human Disease, held by Dr. Jonathan King on February 12, 2015.
Assignment 1 and discussion thereof
For today’s class we read three papers and one retrospective essay on the early history of protein structure [Pauling 1951, Sela 1957, Anfinsen 1961, Perutz 1987]. A useful modern review that connects terms in those papers to what we know today is [Eisenberg 2003].
Pauling, Corey and Branson proposed two energetically stable protein secondary structures [Pauling 1951]. The first (Fig 2 & 4) is left-handed and made of D amino acids, with 3.7 residues per turn and 1.5Å translation along the helical axis per residue. The second is right-handed and made of L amino acids, with 5.1 residues per turn and 0.99 Å of translation per residue. The first structure is very similar to what we now call alpha helices. However, it turns out that in real life, naturally occurring amino acids are all levorotary (L) and alpha helices are right-handed. Also the number of residues per turn is now known to be closer to 3.6. The second structure is now referred to as a gamma helix but is absent from nature. It also turns out there are a few secondary structures which are rarer than alpha helices yet still existent, such as the 310 helix, which Pauling, Corey and Branson did not anticipate.
To this day, no gamma helix has ever been found, even though it looks good on paper. It turns out that an alpha helix has exactly the right geometry such that the backbone atoms are exactly packed to maximize Van der Waals interactions, with absolutely no hollow space. Even though every cartoon of alpha helices ever drawn depicts a hollow core, the core is in fact completely packed solid. In contrast, the gamma helix is wide enough that it would have a hollow core which would fill with solvent. Perhaps this is what makes it unfavorable.
With 3.6 residues per turn and 1.5Å translation per residue, the alpha helix completes one 360° turn every 3.6×1.5 = 5.4Å. This is called the “pitch” of the alpha helix. I have labeled these properties on this PyMOL screenshot of human PrPC residues 146-151 (EDRYYR), a segment of α-helix 1, from an X-ray crystal structure by [Knaus 2001] [PDB# 1I4M]:

To reproduce the above figure, follow these steps in PyMOL (see also this post):
bg_color white
fetch 1i4m
hide everything
show cartoon, resi 146-151
show sticks, resi 146-151
color red, resi 146
color orange, resi 147
color yellow, resi 148
color green, resi 149
color blue, resi 150
color violet, resi 151
A nice overview of supersecondary structures is here. Most βαβ motifs are right-handed, though I always need to look at the diagram to remember exactly what that means spatially.
DTT is one agent to reduce disulfide bonds. Here are the steps by which it acts:
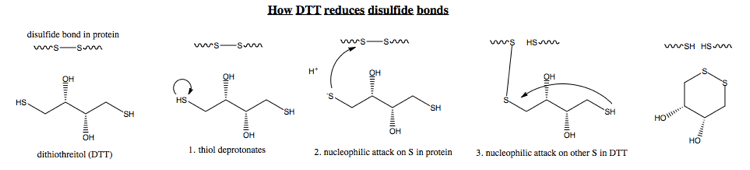
Note that in step 3, I am abbreviating - obviously, the other end of DTT also has to deprotonate before it can do any attacking. Also realize that the other end of DTT also could attack the other cysteine, but the equilibrium lies far in favor of DTT circularizing, because this hexagonal conformation is much more energetically stable. The major difference between the extracellular and cytosolic environments is that the extracellular environment contains oxygen, which favors disulfide bond formation. However, at a fundamental mechanistic level no one understands exactly how oxygen catalyzes this - no one could draw the equivalent of the above diagram but for oxygen.
The mechanism for mercaptoethanol is virtually identical:
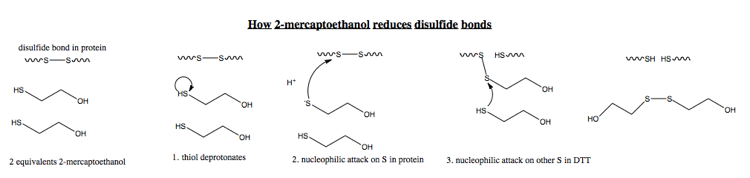
Anfinsen and colleagues discuss five mechanisms (A1, A2, A3, A4 and B) for how proteins might spontaneously fold [Anfinsen 1961]. Here are their conclusions:
- A1. Once one disulfide bond forms in RNAse, the energy barrier to forming the remaining ones is lowered, so there is a tipping point where a molecule will very suddenly adopt the native conformation. They rule out this mechanism because as they re-oxidize the disulfide bonds in RNAse, they are able to free up many thiol (-SH) groups before getting back any enzymatic activity.
- A2. Each of the four unique disulfide bonds in RNAse has a different energy barrier to re-forming, such that as you re-oxidize, first bond 1 will form on every molecule, then bond 2 will form on every molecule, and so on. This would imply that not until you eliminate all thiol groups would you see any enzymatic activity - but in fact they see enzymatic activity earlier, so they rule this out.
- A3. The bonds form in random order. Thus if you had eliminated 3/4 of free thiol, the probability that any one molecule has all eight cysteines oxidized, allowing it to re-form all of its disulfide bridges would be (3/4)8 = 10%. But in fact, they see that when 3/4 of thiol are eliminated, they get >50% enzymatic activity, so they rule this out too.
- A4. The limiting rate for regaining of enzymatic activity is secondary structure folding, not disulfide bond formation. They rule this out on the basis of a number of their earlier studies they cite which suggest that secondary structure formation is very rapid, whereas the lag time in their own experiments is on the order of hours.
- B. Disulfide bonds form incorrectly (meaning between all 8 choose 2 pairs of cysteines rather than only the four correct combinations) at first, and then get rearranged later as folding proceeds. They believe this explanation is most likely.
They had spent a lot of time refining their protocol for re-folding of RNAse. They used very fresh urea because urea, on storage, can degrade into cyanates which will react covalently with proteins, inactivating them. Impurities of thiol reagents can also affect the reactions, so they needed the purest stuff. They also learned to work at the lowest possible protein concentrations, because they found poorer recovery of enzymatic activity at higher concentrations:
| [protein] (mg/mL) | % yield | % recovery after refolding |
|---|---|---|
| 7.0 | 27% | 8% |
| 4.8 | 42% | 29% |
| 2.3 | 87% | 65% |
| 0.9 | 100% | 77% |
| 0.35 | 100% | 94% |
The explanation is that from the reduced, unfolded state, the protein can go down one pathway towards a soluble, enzymatically active monomer, or a different pathway toward an insoluble, aggregated form. Anfinsen and colleagues did study the latter a little bit, and found that the aggregated material had non-native disulfide bonds, and this was a “trapped” state which could be rescued back into a native state via partial denaturation and reduction. This may actually have biological relevance. RNAse probably folds via one pathway when it is coming off the ribosome, but also has to be robust to partial denaturation, which must happen to it often in the stomach. So RNAse probably has the ability to form non-native disulfide bonds, and yet then shuffle them to re-attain native conformation, via a messy pathway that we still haven’t worked out.
Another interesting protein is bovine pancreatic trypsin inhibitor, which is 58 residues and contains 4 disulfide bonds. Though made in the pancreas, it operates in the blood, not the gut. Thomas Creighton studied this and found one particular path of disulfide bond formation - there was a particular S-S bond that always formed first, another that always formed second, and that the optimal folding path required transient formation of a non-native disulfide bond which would bring two halves of the molecule together, at which point other bonds would form and the non-native one would break. This finding was controversial at the time, and indeed, some people still doubt his results even to this day, although the discovery of chaperones helped speed acceptance of the idea that non-native folds or bonds could be on-pathway for native folding.
Lecture notes
The major classes of protein folds are:
- Globular proteins. These are usually soluble, and are the substrate for most biochemistry research.
- Fibrous proteins. These include coiled coil proteins, which we will discuss today.
- Membrane proteins. This is perhaps 30% of our proteome, but they are vastly under-studied because they cannot be crystallized.
- Natively unstructured. These proteins are unstructured in aqueous solution and usually cannot be crystallized. It is unclear whether some of these might actually have well-defined structures in their native compartment of the cell. This class includes alpha synuclein, which we will cover in detail in this class.
Many important insights in the field have come not from computational analysis, but simply through spending a ton of time staring at a structure and contemplating it. For instance, [Chothia 1981] stared at 50 helix-helix contacts in a a number of different proteins, and came to understand the importance of helix-helix packing in defining the fold of globular proteins. The helices they examined were on average 15 residues long (range, 6 to 23) and the interfaces of Van der Waals contact between helices were on average 4.7 residues long (range, 3 to 7). Note that the residues closest in 3D space in these contacts are not necessarily anywhere near each other in 1D sequence, confounding any attempts to gain computational insights.
One of the things Chothia recognized is a topology present on the surface of alpha helices. Grooves and ridges are formed by the i±4n (e.g. 1, 5, 9, 13…), i±3n and i±1n series of residues. The bulkiness of the residues determines the height of ridges and depth of grooves. Pairs of helices usually intercalate 4-4, 3-4 or 1-4, meaning there is almost always at least one i±4n ridge/groove involved.
Helix-helix interface residues are predominantly hydrophobic - 50% are A, I, L, F, V. Another 25% are the hydrophilic D, N, E, Q, K, R, usually found at the edge of the interface, half contacting the solvent.
In helix-helix packing, the distance between the two helix axes avearges 9.4Å (range, 6.8 to 12.0Å), depending on the size of side chains. The interpenetration of atoms from each helix into the other one’s grooves is only 2.3Å, though. In other words, only the tips of the side chains really interdigitate, and the peptide backbone is not involved in the packing at all.
Myoglobins from many different species have been crystallized. It is easy to align the sequences (or structures) due to absolute conservation of the heme-binding histidines. Once these are aligned, you find that only about 15% of the remainder of residues are highly conserved by some metric.
Here, again, is that myoglobin structure from last time:

The fact that almost all proteins use almost all 20 amino acids suggests that each amino acid can do something unique and special at least some of the time. There are amino acids that are often found at the ends of alpha helices and help to define the beginning of a helix - these sometimes form turns and are often called “helix caps”.
Some of the first studies of protein structure used not X-ray crystallography but X-ray fiber diffraction. Fibers of hair diffracted X-rays with meridional signal at 5.15Å and equatorial at 9.8Å. When the hairs were stretched, the pattern changed. This was evidence that (1) keratin in hair had a particular conformation, consistent between all the different copies of keratin, and (2) that this structure could be distorted into a different specific conformation by force. X-ray fiber diffraction has come back into importance in recent years as a tool for understanding amyloid folds.
In contrast to keratin, actin is not fibrous, it is globular, but it polymerizes into filaments. Tropomyosin is a coiled coil fibrous protein which binds actin filaments and walks along them in order to flex a muscle.
Examples of 2-stranded coiled coils include:
- Myosin
- Paramyosin - this is what clamps bivalve sea creatures shut
- M-protein
- Lamin
- Intermediate filament (IF)
- Tropomyosin - introduced above
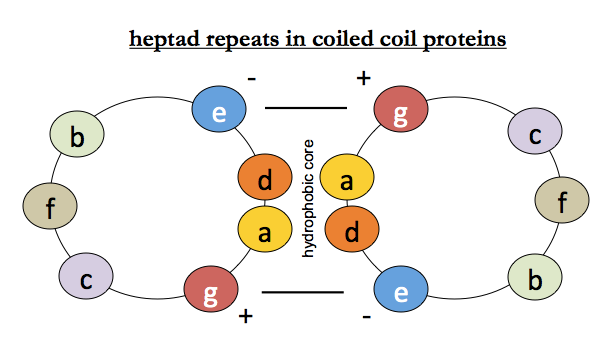
Because you can get large amounts of muscle from animals, it is relatively easy to purify muscle proteins, and their amino acid sequences were determined relatively early on. Tropomyosin, myosin, paramyosin and intermediate filament all contain heptapeptide repeat motifs which are fairly well conserved. Leucine is the most common residue in the 1st (“a”) position of the repeat. Positions a and d are hydrophobic and face into the interface, g and e are charged, and b, c and f face outward and could be anything. This creates a hydrophobic core at the interface, locked together by opposing charges on either helix.
The first complete high resolution structure of a coiled coil protein was that of a fragment of GCN4, a parallel coiled coil with two alpha helices each containing 8 turns and 31 residues [PDB# 2ZTA].
Even though alpha helices themselves are right-handed, they can coil around each other in a left-handed fashion. Helices are said to be parallel if the N termini are both at one end, and the C termini both at the other end, and antiparallel if they are head-to-foot with one helix’s C at the other one’s N. Counterintuitively, both structures are stable and exist in nature. As you might guess from the above diagram, it turns out that the charges of residues g and e on the two strands are a large part of what determines the parallel vs. antiparallelness of the coiled coil.
It turns out there are even 3-stranded parallel coiled coils with axial symmetry, a hydrophobic core and three ionic pairs. Similarly, there are 4-, 5-, and 6-stranded coiled coils. The larger ones are not as tightly packed due to steric issues, but do nevertheless appear in nature.
The coiled coil proteins are considered some of the best examples where we really understand how amino acid sequence determines structure. However, the fact that we can look at the structure and see how sequence helps to hold the protein in its conformation doesn’t actually tell us how the protein got into that conformation in the first place. An analogy is that the wedge shape of stones in an archway is clearly important for the stability of the arch, but it doesn’t tell you how to build the arch. In fact, it is impossible to build an arch without a scaffold - you need the scaffold to support it until you finish it and add the keystone, at which you can remove the scaffold and it will self-support. Moral of the story: the forces that hold a structure together may not be sufficient to create that structure.
Because hemoglobin is soluble and abundant, it was easy to run patient samples on a gel and see a difference in electrophoretic mobility if you had a change in charge. Mutations in hemoglobin were originally named after the cities or hospitals in which they were discovered.
It is interesting to reflect on why others didn’t discover the alpha helical structure before Pauling, Corey and Branson did. Perutz had been trained as a physicist, which may have instilled in him a sense that the universe should be integral. Pauling had been a geologist and was more used to the prosaic randomness of our world. Perutz wasn’t entirely wrong though - it turns out that in order for heptad repeats in coiled coil proteins to work, there needs to be an integer number of residues per repeat unit. In these cases, the helix-helix packing just slightly expands the helix so that it is exactly 3.5 residues per turn, so that an arbitrary number of consecutive 7 amino acid repeating units can interlock.
Note that high-resolution X-ray crystallography data are usually refined computationally before they are published. This refinement can abolish certain subtle things that are not well-understood. For instance we now know that sulfur can form hydrogen bonds and that the distance between cysteine and another side chain is variable depending on the strength of the bond, yet for years, these computational refinements have assigned exactly the same distance between sulfur and another atom.
55 years after the early studies on RNAse, we still don’t understand how the sequence of RNAse dictates its fold, and no one is studying it anymore. An N-terminal alpha helix folds independently in solution, which was useful for determining its structure, yet in the full structure of RNAse, that helix is not independent, but rather packed against another part of the protein. In nature, alpha helices are never found naked - they are always packed together.
We are going to discuss the pathway from a “fully denatured” protein through refolding to a native state, with or without a chaperone. The phrase “fully denatured” might be defined as loss of activity (for enzymes) or loss of antigenicity (if you are probing with antibodies) or a phase change resulting in a visible precipitate. Denaturation is often irreversible, as for the cooking of egg white.
Denaturants include:
- Heat. This is not that useful in the laboratory, because it often causes precipitation, which makes the protein hard to study.
- pH (very high or very low). Also not that useful, because extremes of pH will drive all manner of other chemical changes that will change your protein.
- Organic solvents. For instance, phenol, ethanol or gasoline cause the protein to precipitate.
- Inorganic ions. Lithium bromide, lithium chloride, potassium thiocyanate. Many physiological salts (NaCl for instance) will not denature proteins. NH4SO4 actually stabilizes many proteins, increasing their melting point.
- Detergents. You usually need enough concentration to form micelles, and then the protein inside the micelle will be denatured. A disadvantage is that it is very difficult to subsequently remove the detergent.
- Organic solutes, mainly urea and guanidine hydrochloride, are far and away the most useful denaturants. Urea is non-ionic. Guanidine, which is much more potent, is ionic. Therefore as you add guanidine you are also increasing the osmotic concentration of the solution. Both of these leave proteins soluble in aqueous solution, rather than precipitating them. This is what makes them so useful.
The mechanism by which urea and guanidine denature proteins is not entirely clear. One aspect appears to be that they disrupt the structure of the water around the proteins.
We can categorize protein association reactions as follows:
| name | strength | specificity | reversibile?* |
|---|---|---|---|
| precipitation | weak | non-specific | yes |
| crystallization | weak | specific | yes |
| micelle formation | intermediate | semi-specific | no |
| collagen gelation | intermediate | semi-specific | no |
| aggregation | strong | specific | no |
| self-assembly** | strong | specific | no |
*If you dilute, will it go back into solution?
**Self-assembly refers to things like actin polymerization.
For years, people working in protein production in biotechnology thought that aggregation was the same thing as precipitation. But we now separate these two concepts by an important operational definition: if you add solvent back to it and it does not go back into solution, then it was aggregated, not precipitated. Many small molecules precipitate, but none of them aggregate. Aggregation is a property exclusive to proteins, because it requires a biomolecule large enough to have multiple conformations.
Specific here means that each protein only aggregates with other copies of itself. If you mix two proteins of distinct amino acid sequence and subject them to conditions that will them both to aggregate, they will each aggregate separately [Speed 1996]. This turns out to be an incredibly useful property. When you express a recombinant protein in E. coli, that one protein will aggregate and form inclusion bodies, which do not include any of the thousands of other bacterial proteins in the cell. Now you can take advantage of the insolubility of that aggregate to purify your protein. Like this.
Interesting fact: most cataracts are caused by aggregation, but there is one particular genetic mutation that causes a lens protein to form bona fide crystals in the lens.
Important point for next week
When doing the reading for next week on GCN4, bear in mind that the term “leucine zipper” refers to an outdated and incorrect model of what coiled coils might look like structurally.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.