Prion2015 Day 2

Claudio Soto
Dr. Soto opened with a review of his lab’s development of PMCA over the past 15 years, starting all the way from the beginning [Saborio 2001]. He then focused on recent applications of PMCA to diagnostics. He discussed blood, where his lab achieved detection of prions in hamsters a decade ago [Castilla 2005, Saa 2006]. Last year, his lab was able to amplify PrPSc from urine of 13 out of 14 patients with vCJD [Moda 2014]. Next, he discussed the application of endpoint titration bioassay [Morales 2015] and PMCA [Salvadores 2014] to Aβ. He concluded that section of his talk by expressing hope for the eventual development of a blood-based test for Alzheimer disease, a point which he has made in published literature as well: “adaptation of Aβ-PMCA to detect Aβ oligomers, which may potentially be circulating in the blood of AD patients, may offer a great opportunity for more-routine testing” [Salvadores 2014]. He then presented unpublished results regarding Aβ and blood, which I can’t share here.
Finally, Dr. Soto switched to the topic of environmental routes of prion transmission. He remarked that CWD is readily transmissible in nature, and that this may arise both from direct animal-to-animal transmission and from environmental accumulation due to urine, feces, animal carcasses, etc. depositing prions in water, soil, plants, etc. His lab recently published a battery of experiments on the binding of prions to plants [Pritzkow 2015]. They found, for instance, that plants dipped in prion-infected brain homogenate and then washed retain the ability to infect hamsters by the oral route, and that plants grown in prion-containing soil acquire prion seeding activity detectable by PMCA in their stems and leaves. Last, he discussed some unpublished results regarding prion binding to surfaces.
Q&A
Q. How would the presence of Aβ seeds in blood relate to immunotherapy strategies?
A. David Holtzmann has proposed the “sink hypothesis” [DeMattos 2001], wherein binding peripheral protein seeds with an antibody would result in a net flow of pathogenic seeds out of the brain. Note: an earlier version of this post incorrectly stated that David Housman had proposed this hypothesis. Thanks to Charlie Mays for the correction and citation.
Marc Diamond
Dr. Diamond began by saying that he would present evidence that tau can, for all intents and purposes, be considered a prion.
He reviewed the basic biology of tau. It binds to microtubules, though we still don’t have a full picture of its native function. It contains a repeat domain (RD) which alone is sufficient to form fibrils and cause pathology. There are >25 different tauopathies including sporadic forms, and genetic forms linked to mutations in the MAPT gene.
Following that introduction, Dr. Diamond presented some results he published last year [Sanders & Kaufman 2014] and which his student David Sanders presented in Trieste last year. First, they created cell lines stably expressing different mutant proteins or fragments thereof - huntingtin, Aβ, α-Syn, and Tau RD - and infected them with fibrils made of each type of protein. They observed that “like seeds like” - proteins can seed their own aggregation but not the aggregation of other proteins. In fact, even single point mutations in tau can create transmission barriers between tau prions and incompatible tau substrates. They then did a series of experiments on cell-to-cell transmission of tau prions [Sanders & Kaufman 2014]. This transmission of tau prion seeds appears to be mediated by heparan sulfate proteoglycans (HSPGs) [Holmes 2013], similar to what is known for PrPSc [Horonchik 2005]. Most recently, Dr. Diamond’s lab developed a FRET-based assay to measure tau oligomers in Alzheimer brains, and they estimated that the mininum multimeric size required for a tau prion seed is a trimer [Mirbaha 2015]. When Dr. Diamond presented that result at a meeting recently, Stanley Prusiner pointed out that this result is identical to a result that the Prusiner lab once obtained for PrPSc [Bellinger-Kawahara 1988]. They also used their FRET assay to perform time-series quantification of tau prion seeds in Virginia Lee’s MAPT P301S mice [Holmes & Furman 2014]. He touched briefly on a classic paper on the staging of Alzheimer disease [Braak & Braak 1991], and then returned to the findings of [Sanders & Kaufman 2014], discussing the faithful propagation of distinct tau prion strains in cell culture and upon inoculation back into transgenic animals. He noted their data on different tau prion morphologies in different tauopathy patient brains, and
Q&A
Q. Jean-Philippe Deslys: Didn’t the 1988 Prusiner lab results indicate a minimum PrPSc size of 55 kDa?
A. Marc Diamond: Yes, I re-read the paper carefully last night and have also discussed this with Dr. Prusiner. In that experiment, the PrP was deglycosylated, giving a size of 17-21 kDa, so 55 kDa does indeed correspond to a trimer.
Ina Vorberg
Dr. Vorberg’s talk centered on the intercellular transmission of prions in mammalian cells. She pointed out that the transmission of cytosolic prions between cells requires some means for prions to exit the cell, as well as some means for prions to be taken up into other cells. She noted that Sup35p, like PrP, contains am octapeptide repeat region. In yeast, propagation of Sup35p prions requires Hsp104 [Chernoff 1995], and de novo formation of Sup35p prions requires the [PIN+] prion [Derkatch 2001]. This inspired Dr. Vorberg to fuse a portion of Sup35p to PrP and express it in mammalian cells in an effort to identify analogous mammalian cellular factors involved in the formation and propagation of prions [Krammer 2008a, Krammer 2008b]. They then expressed Sup35NM in mammalian cells and observed self-sustaining propagation and intercellular spread of Sup35NM prions [Hofmann 2013].
The second half of Dr. Vorberg’s talk consisted of new unpublished results on the same theme.
Finally, she recommended two posters on totally different topics by her group, #P.10 and #P.16 - see poster abstracts.
Lary Walker
Dr. Walker covered three topics related to Aβ strains:
- Aβ in humans vs. other primates
- Aβ strains in different patients with Alzheimer disease
- Pittsburgh compound B (PiB), its binding site, and Aβ strains
Primates have very different maximum life expectancies. The record for human longevity is 120 years, while for chimpanzees it is 60 years, and other primates have even lower records. Like humans, many non-human primates develop Aβ plaques as they age [Rosen 2011]. Many of these primates’ brain extracts even transmit Aβ pathology to transgenic rodents, yet Aβ pathology does not seem to result in dementia in any species other than human [Heuer 2012]. PiB is a small molecule probe that binds with high specificity to Aβ plaques in the human brain [Klunk 2004]. Yet, curiously, it does not bind to Aβ plaques in rodents, even when those rodents express human Aβ [Klunk 2005]. There is also essentially no PiB binding in non-human primate brains with Aβ pathology [Rosen 2011]. Dr. Walker hypothesized that this difference in binding may reflect differences in Aβ prion conformation.
Dr. Walker then discussed an unpublished case study.
Next he discussed efforts to identify the PiB binding site [Matveev 2014]. They homogenized and centrifuged human AD brain samples. The supernatant plus “fluffy pellet” contained 95% of total protein and 96% of PiB binding but only 35% of Aβ. The hard pellet contained 65% of Aβ but only 4% of PiB binding. They re-centrifuged the supernatant plus fluffy pellet at 100,000g and obtained an ultra-pellet which contained 5% of total protein but 93% of PiB binding. They then ran this fraction on a sucrose gradient to identify the size of the PiB-binding species. They concluded that the PiB binding site is on a discrete sub-population of Aβ fibrils, and that both proteins and lipids are required for binding. Along with Aβ, they also identified ApoE, tau, ubiquitin, and CLAC (also known as AMY [Soderberg 2003]) as being enriched in the fraction that contained most of the PiB-binding activity. CLAC is interesting because it is not found in Aβ plaques in non-human primates nor in rodents [Matveev 2014]. The presence of lipids in this fraction is also interesting in light of evidence for lipid involvement in Aβ fibrillization [Chi 2008].
Finally, Dr. Walker noted that Aβ shares essentially all of the key prion-like molecular features of PrP, except that it is not known to be transmissible to humans. He and Dr. Jucker recently opined that the term “prion” should be redefined as a backronym for “proteinaceous nucleating particle”, a definition which would focus on the molecular properties of proteins rather than on infectivity per se [Walker & Jucker 2015].
Update 2015-05-29: Dr. Walker’s talk was the subject of a Nature News article.
Q&A
Q. Jean Manson: You describe Alzheimer’s as an exclusively human disease, but cats and dogs have similar neurodegenerative diseases.
A. Lary Walker: Dogs have been extensively studied. They do get Aβ deposits, but like non-human primates, their deposits are mostly vascular, and do not seem to lead to neurofibrillary tangles of tau. It is true that dogs with these Aβ deposits do get dementia, but the disease still may not be sufficiently similar to the human disease to merit being called Alzheimer’s.
Erdem Tamgüney
Dr. Tamgüney presented entirely unpublished work on α-synuclein. Most of the salient details are in his abstract O.01, which is available publicly here, and I received permission from Dr. Tamgüney to post my notes from his talk here.
Dr. Tamgüney’s lab created mice which have endogenous α-Syn knocked out, but have a P1 phage artificial chromosome encoding wild-type human SNCA. These mice express α-Syn at about 1.5x mouse wild-type levels, but have no detectable neuropathology or symptoms within a mouse lifetime. They injected them with brain homogenates from two different MSA patients, or non-demented aged controls (NDACs), or PBS. They then sacked the mice at 3, 6, or 9 months. By 9 months, the MSA-injected mice accumulate phosphorylated α-Syn deposits in the cytosol of neurons. However, so do the mice injected with NDAC brain homogenates. In all of these mice, the α-Syn co-localized with ubiquitin, but not neurofilament light chain (not sure why the latter is significant). They were then able to obtain brain samples for histology from both MSA patients and one of the two NDACs. In the original MSA patients, the phosphorylated α-Syn was found in oligodendrocytes. The one NDAC turned out to have neuronal deposits of phosphorylated α-Syn. This individual may have had early changes preceeding onset of Parkinson disease or dementia with Lewy bodies. Thus, even people without clinical signs of synucleinopathy may have α-Syn seeds in their brains.
Jacob Ayers
Dr. Ayers began with an overview of published in vitro and cell culture evidence for seeding of mutant SOD1, much of which I’ve also reviewed in my protein folding term paper. He also noted his earlier success in transducing the mouse spinal cord with injections of AAV vectors [Ayers 2015]. Based on these findings, he and David Borchelt hypothesized that they could transmit SOD1 misfolding to mice via injections into the spinal cord. They created SOD1 G85R-YFP mice that do not develop spontaneous disease, and found that these mice do develop an ALS-like disease if they are injected with spinal cord homogenates from SOD1 G93A mice [Ayers 2014]. This is the first in vivo evidence for transmissibility of mutant SOD1 aggregates. They also also looked at aged Gurney WT mice [Graffmo 2013], which express high levels of wild-type SOD1 and develop some SOD1 pathology but no clinical symptoms. When spinal cord homogenates from these mice were injected into the G85R-YFP mice, 1 out of 3 mice did develop paralysis. Interestingly, the pathology in those mice looked different than that in the mice injected with G93A homogenates [Ayers 2014], suggestive of possible strain differences. Dr. Ayers then moved into a few unpublished results involving these mouse models. He dubbed SOD1 a “pseudo-prion” in that it has some properties of a prion but is not naturally transmissible and has a “very restricted host specificity”.
Q&A
Q. David Harris: What is missing for SOD1 and for many other “prions” is evidence that the aggregates transmit between cells in vivo. Do you have ideas for how you could demonstrate this for SOD1?
A. Jacob Ayers: It’s definitely an ongoing challenge, and we are working on addressing this.
Ilia Baskakov
Dr. Baskakov opened by arguing that glycosylation remains a poorly understood area of prion biology. PrP is variably glycosylated at residues N181 and N197 (human numbering). There are a wide range of possible glycan chains on PrP depending on linkers (α-2,3 vs. α-2,6 etc.) and the sugars involved (sialic acid, galactose, fucose, xylose, etc). There are exist ~20 sialyltransferases and 4 neuraminidases which help to determine the composition of glycan chains. The variable sialylation of glycan chains is important for self vs. non-self discrimination. Macrophages and dendritic cells recognize “pathogen-associated molecular patterns” (PAMPs) based partly on foreign glycans, and apoptotic cells also exhibit altered glycan composition that helps tag apoptotic bodies for destruction.
It has long been recognized that PrPC and PrPSc each exhibit great variability in glycan composition, but are not categorically different from one another [Rudd 1999]. Dr. Baskakov hypothesized that differences in sialylation between different PrPSc molecules could help to determine their fate.
To test this hypothesis, his lab sought to remove sialylation from PrPSc. The use of bacterial de-sialylation enzymes directly on PrPSc proved inefficient, so they then tried de-sialyating PrPC and subjecting it to PMCA. They found that undersialyated PrPC was the preferred substrate for conversion to PrPSc in PMCA [Katorcha 2014]. Because secondary lymphoid organs are important in peripheral prion spreading [Mabbott 2012], Dr. Baskakov next compared the sialylation profiles of brain- vs. spleen-derived PrPC and found differences [Katorcha 2014]. From here he moved into additional unpublished results regarding sialylation of PrP.
Q&A
Q. Mark Zabel: Have you looked at advanced glycation end products in vivo?
A. Ilia Baskakov: No, we haven’t. Someone else came out with a paper about it last week, though [citation needed].
Q. Byron Caughey: If your theory is correct, how do you explain the infectivity of severely underglycosylated anchorless PrP?
A. Ilia Baskakov: Because macrophages and microglia recognize glycans, we hypothesize there is a difference between undersialyation and the total absence of glycosylation. The latter would make it harder for the immune system to recognize prions.
Bruce Chesebro
Dr. Chesebro presented unpublished work on the very early spread of PrPSc after intracerebral inoculation. His oral abstract O.03 is here.
Jiyan Ma
Unfortunately I missed most of Dr. Ma’s talk because of the long lunch line. When I came in, he was presenting a summary table arguing that de novo prions generated by PMCA from recombinant PrP can possess all the properties of bona fide brain-derived prions, while noting that PMCA can also give rise to self-propagating but non-infectious PrP conformers. These points have been made to some degree in Dr. Ma’s previously published work [Zhang 2013], though it appeared that he was presenting some new data as well.
Fiona Houston
Dr. Houston began with a review of the risks of prion transmission via blood products. Three individuals (all 129MM) have developed vCJD after receiving non-leukodepleted blood transfusions from individuals with vCJD (presumably referring to [Hewitt 2006]), and two individuals (129MV) have developed subclinical infections, one after blood transfusion and one after plasma infusion (not sure what the citation is for this). It is estimated, based on histological analysis of irreversibly anonymized appendix samples, that 1 in 2000 people in Britain carry subclinical peripheral prion infections [Gill 2013]. Therefore, blood transfusion remains an important source of vCJD transmission risk. For this reason, Dr. Houston has been studying blood transfusion transmission of prions, using scrapie-infected sheep as a model system [Houston 2008]. They sought to identify which blood fractions contain prion infectivity, and found that although the blood fractions vary in degree, all of them carry at least some amount of infectivity [McCutcheon 2011]. Dr. Houston’s group also investigated genetic control of sheep susceptibility to BSE prions via oral exposure, and found that the F141L polymorphism significantly altered incubation time [Tan 2012].
Jean-Philippe Deslys
Dr. Deslys presented 10 year followup data on cynomolgus macaques inoculated with a variety of prion strains.
He also reviewed historical data on transmission of different prion strains to non-human primates. For instance, scrapie was transmitted to cynomolgus macaques, but only after repeated passage in mice [Gibbs & Gajdusek 1972], thus leaving doubt as to whether scrapie prions derived directly from sheep could infect macaques. Sheep-derived scrapie prions transmitted disease to squirrel monkeys [Gibbs 1980], but because these are New World monkeys, it left doubt as to whether primates more closely related to humans were susceptible.
He concluded by observing that transmission experiments are limited by the natural lifespan of the host species, and that therefore the results that people see when they attempt to transmit non-PrP “prions” between animals may simply represent the “tip of the pyramid”, where much greater transmissibility might occur if only the animals could be observed for longer.
David Borchelt
Dr. Borchelt began by reviewing the evidence from cell culture and animal models for the growing family of prions - tau, Aβ, SOD1, and so on. Then he recalled the difficulty of transmitting prions from a mouse model of P102L GSS, where prions exhibited no transmissibility to wild-type mice and only limited attack rates when inoculated into hamsters or mice expressing lower levels of the mutant protein [Hsiao 1994]. And the need to delete endogenous MoPrP in order to transmit human prions to mice expressing HuPrP [Telling 1995].
When mice overexpressing wild-type or mutant (A53T) α-Syn (the M20 and M83 mouse models respectively) were inoculated intramuscularly with α-Syn fibrils, they developed a motor phenotype and had reduced survival [Sacino 2014]. Dr. Borchelt also discussed how the transmission of tau prions to MAPT P301L mice is inefficient [Chakrabarty 2015].
Finally, Dr. Borchelt presented the negative results from the SOD1 in vivo transmission paper [Ayers 2014], as a complement to Jacob Ayers having presented the positive data earlier in the day.
Q&A
Q. Something about transgenes.
A. We saw no transmissions of SOD1 aggregates to mice overexpressing wild-type human SOD1. We had them on a wild-type mouse Sod1 background; one could contemplate crossing them to a mouse Sod1 knockout background.
Yury Chernoff
Dr. Chernoff presented unpublished data on properties of Aβ and tau fused to Sup35 fragments and expressed in yeast; you can see his abstract O.06 here. He previously described this experimental system in his Prion2014 talk and today he presented a variety of new data from this system.
Ed Hoover
CWD is now found in free-ranging cervids in 23 U.S. states, with Michigan just yesterday becoming the latest state to report CWD. Dr. Hoover is interested in two questions related to how prions are naturally transmitted in deer: first, which tissues are the earliest to become infected after exposure to prions, and second, how are prions shed from the animal into the environment? Answering these questions requires characterizing the distribution of CWD prions across different tissues in infected cervids. He presented data comparing the diagnostic success of immunohistochemistry, RT-QuIC, and ELISA in lymph nodes of deer [Haley 2014] and using RT-QuIC lag time to quantify prions in different tissues from deer [Henderson 2015]. He also touched briefly on work from Candace Mathiason’s lab which she had presented yesterday at the animal prion workshop.
Qingzhong Kong
Dr. Kong presented unpublished findings on the zoonotic potential of CWD prions. His abstract is not online and I don’t have permission to share any of the results here.
He did open with a backgrounder on CWD, which included the interesting factoid that deer velvet is valued in herbal medicine. Upon Googling it I learned that it’s reputed to be a body building supplement (see this) and, by the name of 鹿茸 (lùróng), is also an ingredient in traditional Chinese medicine.
David Berry
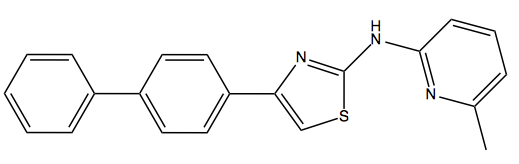
Above: IND24, the Prusiner lab’s lead 2-aminothiazole compound.
Readers who’ve been following antiprion drug discovery efforts will recall that David Berry led the Prusiner lab’s in vivo testing of IND24, its lead 2-aminothiazole compound [Berry 2013]. The compound, which has an EC50 of about 1 μM in cell culture, proved effective at extending survival in mice infected with RML or ME7 prions, but not MM1 or VV2 sCJD prions. The difference in efficacy between RML and MM1 sCJD was observed even when both strains of prion were inoculated into Tg1014 mice [Giles 2010], which express a chimeric mouse/human PrP and are susceptible to prions from both species. This indicated that the specificity of IND24 efficacy was dictated by prion conformation and rather than PrP amino acid sequence.
In that study [Berry 2013] they also found that IND24 was surprisingly effective against one CWD isolate, more than doubling the survival time in transgenic elk PrP mice infected with one CWD prion isolate. When IND24 was administered starting at 1 dpi, the treated mice survived to an average of 237 dpi, while untreated mice only lived to 108 dpi.
Today, Dr. Berry presented unpublished work extending the experiments with IND24 to two additional natural CWD isolates as well as two natural sheep scrapie isolates. His abstract O.07 is here and he gave permission for me to blog his talk.
Dr. Berry tested IND24 against two natural scrapie isolates, SSBP/1 and CH1641, in Tg(OvPrP) mice, and saw absolutely no effect on survival. He then tried CWD isolates from elk and white-tailed deer (WTD) in Tg(ElkPrP) mice and saw about a 2x increase in survival in the elk CWD-inoculated mice and a slightly smaller effect size in the WTD CWD-inoculated mice. The effects were reproducible across a range of IND24 doses from 50 to 210 mg/kg/day.
IND24 treatment alters the strain properties of RML prions, giving rise to prions that have longer incubation times in vivo but are resistant to IND24 treatment in CAD5 cells [Berry 2013]. Therefore, they wondered whether IND24 would also alter the properties of CWD. Surprisingly, though, CWD prions in IND24-treated mice were biochemically and histopathologically indistinguishable from the original CWD prions and retained their original incubation time. They inoculated the original and IND-treated CWD prions into Glenn Telling’s RK13 cells expressing elk PrP, and upon treating the cells with IND24, found no evidence of drug resistance.
Dr. Berry noted that this is exciting news because it means that the drug resistance phenomenon of antiprion compounds may not be inevitable, but rather a property of the original strains being treated. He also noted that it would be interesting to test IND24’s efficacy in cervids, as this would be a way to test the drug discovery model that we are using, wherein we assume that putting an animal’s transgene into a mouse is sufficient to make that mouse predictive of therapeutic efficacy in the original animal.
Finally, Dr. Berry recommended Kurt Giles’s poster #P.121 (see here), which is on medicinal chemistry and dosing regimen improvements for 2-aminothiazoles.
S Jo Moore
Dr. Moore presented her and Justin Greenlee’s work on transmission properties of prions from cattle with the PrP E211K mutation, which is homologous to E200K in humans. This variant was first identified in a cow with atypical (H-type) BSE [Richt & Hall 2008] and later in its offspring [Nicholson 2008]. When they inoculated brain homogenate from an E211K heterozygous animal into another cow of the same genotype, they saw an incubation time of just under 10 months, which is extremely fast for cattle [Greenlee 2012].
They have previously seen early changes in the retina of BSE-infected cattle [Greenlee 2015], so they next asked whether there were retinal changes associated with the E211K H-type BSE. This work is unpublished, but you can see Dr. Moore’s abstract O.08 here.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.