Age of onset in genetic prion disease
Introduction
It may seem like age of onset in genetic prion disease has been studied to death. For the past 25 years, basically every paper to report on a cohort of individuals with genetic prion disease, however large or small, has given at least some basic summary statistics such as the mean and standard deviation of the age of onset or death (see citations throughout this post). Most also include statistics on disease duration, and many have also reported on the significance, or lack thereof, of PRNP codon 129 as a genetic modifier of age of onset. Case series have been reported, reviews written, families counseled.
And to the extent that there do remain some unknowns about age of onset, one might ask, what’s the point in getting more accurate estimates? Even if our current best estimates of the mean age of onset for any given genetic variant are off by a year or two, the variance is so great anyway, with standard deviations on the order of 5 or 10 years, that the difference in prognosis for any one individual being counseled ends up being negligible.
This post will present my counter-argument that there is actually still a lot we don’t know, and that it matters.
What is it don’t we know? Here’s a quick rundown of points I will make in this post. Simply put, we don’t know what the true distribution of ages of onset looks like. Published estimates of age of onset appear to have risen over time, probably due to more thorough ascertainment, and it is not clear if they have now stabilized or still have further to go. We also don’t know how a patient’s current age affects their expected age of onset. The kind of actuarial models that could give us insight into that relationship have not been built. And we also don’t know for which genetic variants the patient’s codon 129 genotype affects age of onset or disease duration, and we don’t know whether age of onset tracks within families.
And why does it matter? There are two reasons why age of onset is important: genetic counseling and clinical trial design. In genetic counseling, some might argue that descriptive statistics about age of onset aren’t useful to patients anyway, since there is so much variance and everyone is unique. Personally, I am a believer that more information is better. But even if you disagree, I will give examples to illustrate that some assertions for which there’s currently no evidence in the literature are already being used clinically, and if doctors are going to give information anyway, we’d certainly like it to be the right information. Meanwhile, for clinical trials, having the right model for age of onset influences who you choose to include in trials, and how outcomes are measured.
A clinical trial of prophylactic doxycycline in asymptomatic D178N-129M individuals (EudraCT 2010-022233-28) is currently using age of onset as its primary endpoint for efficacy [Forloni 2015]. I don’t look to that trial as a model, for two reasons. First, I don’t think doxycycline works. Second, even for a therapeutic I was optimistic about, I would not be content to wait until 2022 to find out whether it’s effective. And even if I was content to wait, funding such a trial (just being realistic about how drugs are made) would probably be impossible, given that the trial would last most of the life of a pharmaceutical patent. Nonetheless, while I don’t think that age of onset is viable as a primary endpoint in a Phase 2/3 trial, it’s hard to get away from age of onset entirely. Even imagining an optimistic scenario where we discover a biomarker in asymptomatic individuals that predicts progression towards symptom onset, and supposing that we manage to get Accelerated Approval from the FDA based on improvements in that biomarker in a Phase 2/3, we would still be expected to show an effect on a bona fide clinical outcome in a Phase 4 trial in subsequent years after approval, and that would probably mean showing that onset of disease is delayed in those who take the drug. I’m not making this up. Over the past two years Sonia and I have been pounding the pavement both in Cambridge, Massachusetts and at conferences all over the country, talking to people from pharmaceutical companies and asking what it would take to get them interested in working on our disease. One thing I’ve heard over and over again (along with biomarkers, and a way to recruit patients for trials) is that we need to gather the best possible data on the natural history of genetic prion disease, including age of onset and disease duration.
Age of onset over time
When I looked in the literature for reported estimates of central tendency for the age of onset in E200K patients, here’s what I found:
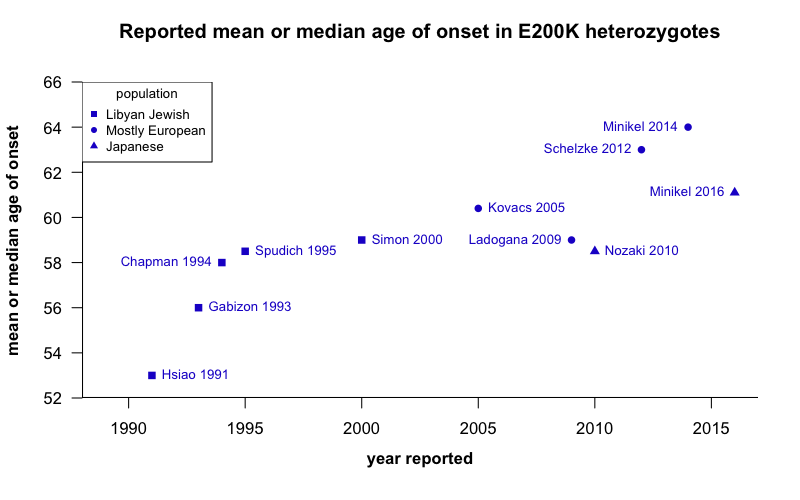
code. Note that “heterozygous” here refers to the E200K mutation itself and not to codon 129. Links to references from above image: [Hsiao 1991, Gabizon 1993, Chapman 1994, Spudich 1995, Simon 2000, Kovacs 2005, Nozaki 2010, Schelzke 2012, Minikel 2014, Minikel 2016]
Reading the literature, I kept getting the impression that there was an upward trend over time, and this plot probably gives you that impression too. Whether this is even a real trend in the literature, I don’t know: my review of the literature here is non-exhaustive, I haven’t done any statistical tests (earlier studies had smaller sample sizes, so the age of onset may not actually be significantly lower than it is today). Also, I have arguably committed a data visualization atrocity by breaking the y axis, thus making the differences look larger than they are. But I wanted to make the point that if you look to the literature to find out what the typical age of onset is, the answer could vary a meaningful amount — by over 10 years.
And there is no reason to think that the actual age of onset has been changing — these diseases unfortunately remain as untreatable as they have ever been. Instead, I see at least three forces that probably explain most of the variation between these different studies:
- Ascertainment. The cases that are exceptional enough to come to the attention of the biomedical research establishment early on might be earlier-onset cases. Over the past few decades, prion disease has become better understood, doctors have become more aware of it, diagnostic tools have improved, and surveillance has improved. All of these factors probably make it more likely that more elderly patients will be correctly diagnosed, rather than mislabeled as Alzheimer’s or something else.
- Different models and inclusion criteria. The studies cited above differ a lot in which patients included in the statistics. Some studies include only definite and probable cases, meaning people who got sick, and were observed directly by the researchers. Other studies also include suspected or assumed cases culled from family histories. And yet others have constructed survival curves that account for people known to still be asymptomatic at the time of the study. To the extent that these mutations are not quite 100% penetrant, and/or elderly cases are not necessarily always diagnosed correctly at the time of death, including family histories and accounting for asymptomatic people will raise the estimated age of onset. Which relates to the third point…
- Predictive testing. People who aren’t sick yet can only contribute increasing the estimated age of onset if their genetic status is known (at least to researchers if not to themselves). Although early papers did often include asymptomatic relatives in analyses [Hsiao 1991], it has never been clear to me what proportion of potentially at-risk people were being ascertained. The only published data I’m aware of on uptake of predictive testing come from the U.K. [Owen 2014], where it appears that the rate of predictive testing has increased quite a bit over time (see Figure 1).
So, to be clear, the plot shown above is absolutely comparing apples to oranges, both in terms of which patients are included and how the data are analyzed (and also in terms of PRNP haplotypes and populations, though we don’t know if these matter). And that is exactly my point: our best estimate of age of onset depends on which data we look at, and how we ask the question.
So how to do it right? As others have observed, it is currently impossible to assess age of onset in genetic diseases the right way [Langbehn 2010]. The truly correct way to assess age of onset (and penetrance) would be to genotype many millions of people at birth, find those who have pathogenic PRNP variants in their genomes, and then follow those people for 90 years and see whether and when they die of prion disease. Obviously, this kind of prospective study has never been done for any genetic disease, since the technology to identify the variants that cause genetic diseases was only invented 30 years ago. But as I’ll argue at the end of this post, even with the data that exist today, it should be possible to do better than we’ve done so far.
A year older, 9 months closer to death
I love the dark humor of those birthday cards that say “Happy birthday! You’re a year older, and a year closer to death.” But as any actuary knows, and as I’ve touched on in this post, that’s not actually how it works. On your birthday, you’ve aged a year, yes, but you’ve also avoided all the things that might have killed you in that year, and that’s worth something. For instance, according to the U.S. Social Security Adminstration’s 2011 Actuarial Life Tables, an American female at birth has a life expectancy of about 81, but by age 70, her life expectancy has risen to 86 and 4 months. By her 71st birthday, her life expectancy rises further to 86 and 7 months. Thus, she’s a year older, but only 9 months closer to death.
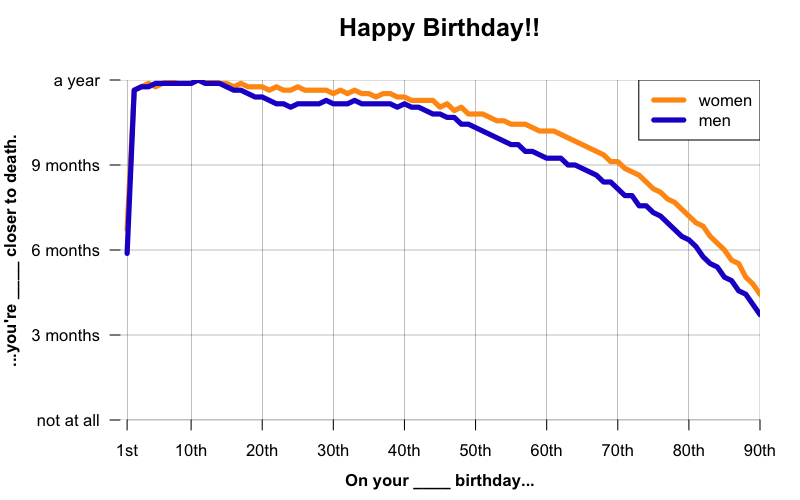
The same phenomenon occurs in genetic diseases. When people say that the D178N-129M haplotype has an average age of onset of, let’s say, 49, that’s averaging across a huge range (from 20 to 72 in one review [Kong 2004]). Obviously, if someone is 50 years old, it can’t be the case that they on average have -1 years left to live. Similarly, someone who is already 45 probably has, on average, more than 4 years left to live.
For prion disease, I am not aware of any published models showing how expected age of onset varies as a function of a person’s current age. Similar models have been constructed for other diseases, though, such as Huntington’s disease [Langbehn 2004, Langbehn 2010]. Just as you’d expect, the models indicate that the longer you’ve already stayed healthy, the later your onset is likely to be. As one example, their models estimate that a person at risk for Huntington’s disease, with 42 CAG repeats, as of age 40 has an 80% chance of onset by age 60, but if they’re still healthy at age 55, they have only a 50% chance of onset by age 60 [Langbehn 2004, supplementary life tables].
Developing a credible model for how expected onset varies as a function of current age will be important for doing clinical trials in genetic prion disease. Again, as noted in the introduction, I don’t believe that age of onset is a good primary outcome for a clinical trial, but even if we find a biomarker we can use in Phase 2/3 trials, I do think that age of onset may be needed for monitoring long-term efficacy in a Phase 4 trial. Moreover, the process of discovering and validating such a biomarker in the first place would benefit greatly from a good model of age of onset, and calculating statistical power and choosing the patient populations for a trial may require it too.
Genetic modifiers
Here’s what I have been able to find in the literature about the effects of codon 129 and of historical age of onset within a family:
| pathogenic variant | codon 129 het vs. hom affects age of onset? | codon 129 het vs. hom affects disease duration? | parent and child age of onset correlated? |
|---|---|---|---|
| 6-OPRI | Yes (10 years) [Baker 1991, Mead 2006] | No [Mead 2006] | Yes [Webb 2009] |
| P102L | No [Kovacs 2005], Marginal [Mead 2006], or Yes (7 years) [Webb 2008] | No [Webb 2008] | Yes [Webb 2009] |
| A117V | No [Kovacs 2005] | ? | Yes [Webb 2009] |
| D178N-129M | No [Goldfarb 1992, Kong 2004, Kovacs 2005, Mead 2006] | Yes [Kong 2004] | ? |
| E200K | No [Gabizon 1993, Kovacs 2005, Mead 2006, Minikel 2014] | Yes [Mitrova & Belay 2002, Minikel 2014] | No [Minikel 2014] |
As the table shows, the currently available evidence gives different answers for different genetic variants. As of now, I think we don’t know to what extent this reflects true biological differences between variants, versus simply reflecting a lack of statistical power for some variants.
I believe there is pervasive confusion about the contents of the above table. When Sonia and I first received our genetic test result (she’s D178N cis-129M, with 129V in trans), a staff member at the National Prion Disease Pathology Surveillance Center in Cleveland told us that it should be at least some consolation to us that, although she had inherited the pathogenic D178N variant from her mother, at least Sonia’s codon 129 genotype was heterozygous (MV), which meant she might have a relatively later age of onset. That staff member has since left the surveillance center, and in the four years since then, I have yet to find a reference in the literature to support that claim. Instead, every paper I’ve read (see above table) has failed to find any evidence that the trans codon 129 affects age of onset for D178N-129M individuals. At conferences over the past few years, I’ve spoken to neurologists who have professed to counseling patients that a 129MV genotype foretells later onset for E200K individuals too (again, I have found no evidence of this in the literature). I have also met doctors who tell patients that their deceased parent’s age of onset is the best predictor of their own future age of onset, even though there is no evidence that parent and child age of onset are correlated for D178N or for E200K, and moreover, I and colleagues have found that parent-child age of onset correlation can sometimes just be a statistical artifact anyway [Minikel 2014].
What we can do
I hope that this post has convinced you that:
- There exist in the literature a wide range of estimates of mean or median age of onset, probably because of different study designs and inclusion criteria.
- Expected age of onset should depend upon a patient’s current age, and we don’t yet have any good models of this relationship.
- There remain unknowns and misperceptions about codon 129 as a modifier of genetic prion disease.
As I’ve argued above, the “right” way to study age of onset is prospectively, by following people from birth. But we don’t have 90 years to wait to do this the right way. So here are some ways in which I think we can use the currently available, largely retrospective, data, to improve upon the status quo.
First, we can get bigger numbers now. Some surveillance centers have reported age of onset statistics before [Kovacs 2005, Nozaki 2010], but the data have never been aggregated worldwide, and thanks to continual improvements in surveillance, the number of cases observed has risen dramatically in the few years since those studies were published. In our recent study of penetrance [Minikel 2016], there were a total of 1,895 individuals with definite or probable prion disease who had rare PRNP variants. That includes some variants of low or unknown penetrance, as well as over 1,000 of those individuals with one of the three most common, highly penetrant variants — P102L, D178N, and E200K — and over 100 people with octapeptide repeat insertions. Aggregating age of onset data for as many people as possible will offer the greatest statistical power to dissect the impact of genetic modifiers.
And if codon 129 still proves not to be a modifer for some mutations, I am also interested in running power simulations to see what effect size we can rule out. While one can never prove the negative, one can use simulations to suggest the negative. Specifically, if with larger datasets we still find that there is still no evidence that codon 129 genotype affects age of onset for E200K or D178N-129M, we can use simulations to ask what effect size we should have been able to detect. For example, in one possible simulation, by randomly permuting the observed ages of onset and codon 129 genotypes for E200K individuals, and then adding 1 (or 2, or 3, etc.) to the age of onset for all MV individuals and testing for a significant difference, we could ask, “What is the largest true effect that codon 129 could have on age of onset and yet remain undetected at our current sample size?”
Finally, and most importantly, I want to do actuarial modeling. Through the use of age data from people who are currently alive and well or who died of causes unrelated to prion disease, we can build more accurate survival curves, and this will allow us to estimate expected age of onset conditional on a patient’s current age, adjusted for any genetic modifiers as applicable. There are still great challenges here — as noted above, only a minority of people at risk undergo predictive testing, and it is often impossible to determine whether people in the family tree who died of other causes might have harbored a mutation and never gotten sick. At a minimum, including the asymptomatic individuals for whom we do have data will be an improvement over using only data on individuals with disease. Clinical centers (including both surveillance centers in some countries, as well as other clinical investigators) that follow families over time will have follow-up data on individuals who underwent predictive testing years ago, and will have pedigree data that will allow identification of obligate carriers.
I am also contemplating what clever tricks we can do to estimate how biased or how limited our ascertainment still is, and try to correct for it. For instance, by looking at people who have undergone predictive testing and then been followed prospectively for at least a few years, we can ask how their hazard (risk of developing disease each year) compares to people assessed retrospectively in diagnostic tests. By looking at individuals in pedigrees who (in hindsight) had prion disease but were never diagnosed in their lifetime, as well as by asking whether there is indeed an upward trend in mean age over time (as the plot at top suggests), we may be able to estimate how much our ascertainment is still skewed toward the relatively younger cases. Of course, I can also imagine all manner of potential biases that will confound interpretation of these analyses, so I am continuing to brainstorm what are the best questions to ask, and I welcome your input.
Indeed, I’m blogging all of this because I am interested in feedback from readers. How can the study design here be improved? What clever actuarial tricks can be used to account for the fact that our data will still, inevitably, be incomplete?
And to those of you who run either surveillance centers, clinical studies, or both, are you interested in collaborating with me on this?
To patients interested in contributing their own and/or their family’s data: weirdly, I can’t accept your data directly. At least not as of today. I am not a clinical researcher, and I do not currently have an ethical protocol in place with my institution that would permit me to do research on data that I’ve gathered directly from patients. Instead, I can only analyze such data if it is collected by other researchers, de-identified, and then shared with me. This hasn’t been much of a problem so far, because other researchers in the prion field have been generous about sharing data, so for now the best way to contribute to this effort is to share your data with another researcher. Please feel free to ping me for more details.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.