Why doxycycline's failure in CJD clinical trials is no surprise
Last month, clinical research teams from France and Italy jointly reported on the long-awaited results of double-blind clinical trials of doxycycline for sporadic Creutzfeldt-Jakob disease [Haik & Marcon 2014]. The result in two words: no efficacy. But before I dissect the results of this particular study further, it’s worth a lengthy digression to explain some history: how did doxycycline, originally an antibiotic, come to be tested in a clinical trial for prion disease in the first place?
I narrated much of backstory in a post in 2012, but that was when I was just getting started at this. Now, with the perspective of another year and a half of reading and blogging about prion disease therapeutics, my read of the literature has changed, and I think this subject is worth revisiting.
The story begins with an anti-cancer chemo drug candidate called 4′-iodo-4′-deoxydoxorubicin (IDOX for short). IDOX was intended as a replacement for doxorubicin (Doxil), a very old chemo drug used for several blood cancers, including myeloma. Myeloma is often associated with AL amyloidosis, a condition where the light chains of antibodies in the blood aggregate into amyloid fibrils. In treating myeloma patients with IDOX in the mid-1990s, one Italian research group noticed, apparently serendipitously, that IDOX didn’t just fight myeloma, it also seemed to help alleviate the clinical signs of amyloidosis [Gianni 1995 (ft)]. Further investigation revealed that IDOX could bind not only to AL amyloid fibrils, but also to several other types of amyloid, including amyloid beta and transthyretin, all apparently with picomolar affinity (kD reported as 5.9e-11 M) [Merlini 1995].
Along the way, IDOX also underwent a sort of testing for efficacy against prion disease too [Tagliavini 1997]. I say “sort of” because the experimental design, though technically in vivo, cannot really be called a therapeutic study. Control hamsters were infected with scrapie brain homogenate, and another group of hamsters were infected with scrapie brain homogenate that had been co-incubated with IDOX before injection. The co-incubation was found to reduce the infectious titer of the prions, making those hamsters live longer.
In 1997, this work was published in Science; today, you’d be lucky to get a study of this experimental design published at all. I say that not so much as a criticism of the paper, but as a note of how incredibly far the prion field has advanced in the past 17 years. Plenty of different compounds are now known to reduce infectivity in brain homogenate, but that doesn’t make them all anti-prion drugs. If you wanted to disinfect an infected substance, then bleach, heat or acid would do just as well. Making a drug that works in vivo is much harder, and the sort of “co-incubation” experiments used in Tagliavini’s Science paper and many other prion studies around the same time just don’t demonstrate in vivo efficacy. The experiment that does demonstrate in vivo efficacy is to infect animals, wait a while, and then treat them with the drug. That was never done with anthracyclines.
IDOX eventually underwent a clinical trial for primary systemic amyloidosis but, as far as I can tell, it never made it to FDA approval for any indication. IDOX was probably limited by the same problem that Tagliavini cited as his reason for not doing a true in vivo treatment experiment: it’s seriously very toxic. IDOX and doxorubicin belong to a class of drugs called anthracyclines which are known to cause cardiac arrhythmia and cardiomyopathy. This motivated the question of whether less toxic, structurally similar molecules might have the same effect. As it happens, anthracyclines have a 4-cycle core structure somewhat reminiscent of that of a class of antibiotics called tetracyclines.
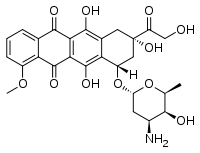 Doxorubicin, an anthracycline chemo drug.
Doxorubicin, an anthracycline chemo drug.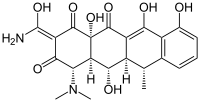 Doxycycline, a tetracycline antibiotic.
Doxycycline, a tetracycline antibiotic.Tetracyclines therefore started to seem like potential treatments for prion disease. Tagliavini conducted a series of in vitro experiments on tetracycline, the original drug in on which the tetracycline class of drugs was based [Tagliavini 2000]. It was found to bind to PrPSc fibrils and revert their protease resistance, and it also bound to PrP106-126 peptides – the latter being a synthetic peptide corresponding to a highly conserved amyloidogenic region at the center of PrP. PrP106-126 peptides are known to kill neurons in vitro [Forloni 1993], but apparently not when bound by tetracycline [Tagliavini 2000]. This work confirmed that tetracyclines are capable of binding PrP amyloid much like anthracyclines It accomplished a couple other cool things as well – it established a binding site on PrP106-126, and in testing whether tetracycline could revert protease resistance of PrPSc it did a full dose-response from 20 nM to 2 mM for two different human prion strains (type 1 and 2 sCJD). Tagliavini was a collaborator on another study a few years later which confirmed these results (again with type 1 sCJD) and also showed that tetracycline inhibited PMCA with 263K hamster prions [Barret 2003]. Both studies left one important question unanswered, though. Cell culture models of prion infection had already been in use for several years and the anti-prion behavior of some molecules like Congo Red had been discovered in these cell models [Caughey 1992], yet neither study tested tetracycline in prion-infected cells.
By now, we know what the answer would have been. FDA-approved drugs, which include multiple tetracyclines, have been screened for antiprion drugs a few times [Kocisko 2003, Poncet-Montange 2011, Silber 2013], and tetracyclines have never once been reported as a hit, suggesting they probably don’t reduce prion accumulation in cultured cells.
Within a couple years of Tagliavini’s in vitro study, his group published the results of a co-incubation experiment with tetracycline, repeating almost exactly the same study that he’d done in 1997, but swapping tetracycline in place of IDOX [Forloni 2002]. The results, too, were similar: animals injected with scrapie brain homogenate lived longer if the brain homogenate had been co-incubated with tetracycline. This experiment troubles me: the justification for conducting a co-incubation experiment with IDOX had been its toxicity, but tetracyclines are approved drugs with excellent safety profiles. It is not clear why the authors chose not to test tetracycline in a true in vivo therapeutic administration experiment.
Such an experiment was eventually done, six years later, and still not up to the highest standard of evidence [De Luigi 2008]. Hamsters were infected by a variety of peripheral routes – subcutaneous, intraperitoneal, intramuscular – and were then immediately (within one hour, at the same injection site) treated with tetracycline, doxycycline or minocycline. Sure enough, the drugs delayed onset of disease by 25 to 81%.
But this is almost like a co-incubation experiment, except that the co-incubation occurs after injection into the animal instead of before. Injection at the same site within one hour might leave an opportunity to “treat” the disease by simply inactivating some of the original inoculum rather than by preventing new PrPSc formation. Moreover, it’s always easier to treat prion infection in the periphery than in the brain – in fact, by the time De Luigi’s study came out, peripheral prion infections could already be completely cured by antibodies [White 2003] or considerably delayed (~3x) by phthalocyanine tetrasulfonate [Priola 2000]. So by 2008, the problem wasn’t that we don’t know how to treat peripheral prion infection, it was that people don’t get peripheral prion infections. They get brain infections. As far as we can guess, most prion infections probably start in the brain, and in any case, by the time anyone is diagnosed, the infection has to have been in the brain long enough to cause serious symptoms.
Accordingly, De Luigi also did some experiments in which animals were infected intracerebrally and then treated 30 or 60 days later with liposome-entrapped tetracyclines infused directly into the brain. The survival index in these experiments was considerably lower than in the peripheral infection experiments: treated animals lived 8 to 10% longer than untreated ones, with p values ranging from .02 to .06. The problem with this result is not that patients wouldn’t like to live 10% longer – many certainly would – it’s that it can be very hard to tell if such small differences observed in one experiment are even real or not. There are many reasons to treat such scientific findings with a bit of healthy skepticism [Ioannidis 2005].
And mind you, even if those results are real, they don’t reflect a realistic treatment scenario in the clinic. The study claims that the animals treated at 60 days already showed symptoms of prion infection – “hyperreactivity to tactile and acoustic stimulation” (data not shown). If true, this would be by far the earliest anyone has been able to observe symptoms of prion disease. The control animals died at 130 days, so 60 days is timepoint 60/130 = 0.46. As explained here, the earliest reported symptoms in any other model are at timepoint ~0.67 [Mallucci 2007]. Moreover, as I’ve discussed regarding pentosan polysulfate, the reality is that sCJD patients don’t exactly join a trial the moment they have symptoms – indeed, the mean time from onset to diagnosis is reported to be 8 months [Paterson 2012].
If one really wants to show that an antiprion drug is a solid candidate for further trials, then one sound experimental design is as follows: infect animals intracerebrally, then treat them orally or intravenously with the drug starting at a few different timepoints – say, 1 day after infection, 50% of the way through the disease, and 75% of the way through the disease. Indeed, by 2008, one small molecule, cpd-b, had already been shown to be effective against prion infection under these rigorous conditions [Kawasaki 2007].
Around the same time as De Luigi’s study, another group published results on minocycline, another tetracycline drug, under a similarly rigorous experimental design [Riemer 2008]. Mice were infected intracerebrally with 139A prions and treated orally, from 100 days post-infection (timepoint 100/178 = 0.56), with minocycline. There was no difference in survival (p > .05, n = 17 vs. 16).
I’ll sum up all this data on tetracyclines from preclinical animal trials as follows: tetracyclines reduce the infectivity of injected prions, but their effects once infection has taken hold in the brain are marginal at best, and quite possibly non-existent. Yet it was on the basis of this level of evidence that tetracyclines moved forward to clinical trials.
By the way, tetracyclines have been pushed forward for more than just prion disease: minocycline has already made it to human clinical trials for practically every other neurological condition you can imagine: Fragile X, ALS, autism, Huntingtons, Alzheimers, Parkinsons, and yet others. The mostly negative results from these trials have earned tetracyclines a weary reaction from neuroscientists. The skepticism is such that when I told a Huntington’s researcher last fall that Prion Alliance was crowdfunding a preclinical trial of a drug candidate, he said, “I hope it’s something better than doxycycline.”
By the time De Luigi’s study was published in 2008, it was stated that Tagliavini’s group had already been treating CJD patients with doxycycline under compassionate use for five years [De Luigi 2008]. Other groups had done so as well, and results trickled out to the public in the form of a sentence or a paragraph here and there in a review article [Tagliavini 2008, Stewart 2008, Zerr 2009] and eventually in the form of conference presentations, but by the end of 2013 – a full decade after Tagliavini started treating patients – there was still not a single publication specifically addressing the efficacy of doxycycline for Creutzfeldt-Jakob disease.
At last, we now have that, although sadly the full text is paywalled [Haik & Marcon 2014]. The study reports the results of a double-blind, placebo-controlled trial run in France by Jean-Philippe Brandel and in Italy by Fabrizio Tagliavini. The study design was based in part on projections from open label trial results. It is implied that patients who chose doxycycline in those open-label trials lived an average of around 11 months, while patients who declined doxycycline lived about 6 months. The placebo-controlled trial was designed to have 80% power at p = .05 to detect this magnitude of effect, so a ~2x increase in survival time after randomization (not a ton of power, but this is after all a rare disease). The study randomized 121 patients, and when the data were analyzed, whether all together or stratified by age, sex, genotype and so on, there was not the slightest whiff of a difference in survival between treated and control patients.
We shouldn’t be surprised by this. Doxycycline was effective at reducing the infectivity of injected prions, but its efficacy in treating a really late-stage prion infection was never evaluated in an animal model. There are lots of drugs that work in animal models but subsequently fail in clinical trials, while I have yet to hear of the converse. To take a drug to clinical trials which has not shown efficacy in an animal model comparable to the treatment scenario envisioned for patients is to invite failure.
Is there another treatment scenario in which doxycycline could work for patients? The authors note that a clinical trial of doxycycline in presymptomatic fatal familial insomnia carriers is ongoing in Italy (EudraCT 2010-022233-28). I’ve met several PRNP mutation carriers in the United States who are taking doxycycline off-label in the hopes this will prove to work. The trouble with the presymptomatic trial for FFI is that it is scheduled to run until 2022. That’s an eternity. By 2022, I sincerely hope that we’ll have much more promising drug candidates than doxycycline to administer to presymptomatic mutation carriers.
If we really wanted to know whether doxycycline will be effective in treating presymptomatic mutation carriers, we could first test that question in animals. We now have mouse models of FFI and other genetic prion diseases which get sick in under a year – a lot quicker than waiting until 2022 [Jackson 2009, Yang 2009, Jackson 2013]. In fact, when we at Prion Alliance launched our fundraising campaign to test anle138b against genetic prion disease last year, we considered testing doxycycline alongside anle138b. But in the end, after several discussions, we decided we couldn’t justify spending the money because we just didn’t think doxycycline looked promising enough.
Why not? Because lots of drugs have been shown, in at least one study, to extend survival in a prion-infected animal by 5 or 10%. Lots of drugs have been shown to have prophylactic effects if administered before prion infection or co-incubated with inoculum. When we objectively compared the standards of evidence for various drugs being effective, there was no obvious reason why doxycycline should float to the top. This is true even when one restricts the search to FDA-approved drugs with excellent safety profiles for long-term use: the evidence for statins as having therapeutic effects in prion infection is far stronger than for doxycycline.
I believe that understanding all of this backstory is incredibly important. By taking the full view of how drugs reach a clinical trial and understanding why those clinical trials fail, we can know what to do right in the future and, equally important, we can know where to place our hope and our optimism.
Though several drugs have been tested here and there in a handful of patients, at this point basically four drugs have been tested in humans with prion disease in some sort of systematic way: flupirtine [Otto 2004], pentosan polysulfate [Bone 2008, Tsuboi 2009], quinacrine [Collinge 2009, Geschwind 2013], and now doxycycline [Haik & Marcon 2014]. I submit that the failure of all four of these drugs to extend survival could have been handily predicted beforehand based on the poor standard of evidence for efficacy going into the trials (see posts on quinacrine, pentosan polysulfate, and a brief discussion of flupirtine here).
Whether that should have deterred us from even attempting such trials can be debated. Patients have no other options, and may want trials even if the probability of success is low. Meanwhile, the world has actually gained some valuable institutional knowledge of how to run clinical trials for prion disease through running these failed trials. On the other side of the scale are the enormous cost of running clinical trials, and the intangible cost of patients getting their hopes up, only to have them dashed again.
That latter cost is the one I want this post to address. Doxycycline clinical trials didn’t fail because prion diseases are inherently untreatable or impossible to cure. They failed because doxycycline-for-prion-disease wasn’t that well vetted of an idea in the first place We can do better, and we will.
Consider just how far prion science has come since the doxycycline story began in the mid-1990s. Back then, it was an open question whether prion disease even could be inhibited by small molecule drugs – the only compounds known effective in vivo were monstrous large molecules like amphotericin B and pentosan polysulfate. An FDA-approved drug that could so much as reduce prion infectivity in brain homogenate in a test tube was big news. It wasn’t until 2007 that cpd-b came along and demonstrated that finding an effective antiprion small molecule could even be feasible. And now, in just the past year, we’ve seen in vivo results for two more such molecules announced - anle138b and IND24. These molecules weren’t discovered through serendipity, they were discovered through systematic high-throughput screening. When molecules like these fail – and many will, because the standard they’re being held to is vastly higher than any test doxycycline ever had to pass – there are dozens of other compounds ready to take their place, coming off of the pipelines of drug discovery efforts around the world.
In the whole chemical universe of compounds you could explore in search of a treatment for your disease, the odds of finding a really effective molecule that also just happens to already be an FDA-approved drug are pretty poor. The odds of any one clinical trial succeeding are never great, and are especially bad if the trial is undertaken without conducting the most rigorous possible animal studies beforehand. If doxycycline had turned out to work, it would have been an unbelievable stroke of luck. But we have something better than luck: science. Today may a bad day for doxycycline, but it’s a fantastic day for optimism.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.