Why we want to test anle138b in GSS mice
Earlier this week we at Prion Alliance launched a Microryza fundraising campaign for our first therapeutic study, to test anle138b in GSS mice expressing the equivalent of the A117V mutation in humans. Here’s why.
why therapeutic trials in genetic prion disease mice?
Over a hundred experimental treatments for prion disease have been tested in animal models. There are two main things we hope to add to this body of research in order to move the field toward meaningful therapy for human use.
First, most of the therapeutic trials to date – all but two [Cortes 2012 (ft), Canello 2012] – have been in mice infected with prions exogenously. Such studies use rodent prion strains that are certainly a useful model of human prion disease, but do have strain-specific properties that mean therapeutics won’t always translate to human strains, as demonstrated by the 2-aminothiazoles. Mice carrying Prnp mutations from human genetic prion diseases aren’t a perfect model either – nothing is – but at least, these mouse models often manage, with a single amino acid substitution, to recapitulate the biochemical and phenotypic properties of the human disease caused by the corresponding substitution in PRNP. That’s a clue that these mice are producing the same strain of prion – meaning same conformation of protein – or at least something very close. Therefore we are hopeful that therapeutic trials in such mice will have direct relevance to treating humans with these mutations.
Second, though we don’t yet have a drug candidate that is highly effective very late in the course of prion disease, several compounds have shown stronger efficacy when administered early in prion infection, such as at 0 or 1 dpi. That’s not a realistic treatment scenario for sporadic Creutzfeldt-Jakob Disease, where 2/3 of the symptomatic phase of disease passes before diagnosis is reached [Paterson 2012 (ft)]. But it is a realistic treatment scenario for carriers of genetic prion diseases, who may know their gene status for decades without encountering symptoms. Mouse models of genetic prion diseases share with human genetic prion diseases the feature that infectivity arises spontaneously, from zero titer, which at best could mean that therapeutics will have an even larger effect in these diseases than RML-infected mouse models would suggest. Only experimentation will tell. To be clear, novel experimental compounds (such as anle138b) will need to pass clinical trials in symptomatic patients before any doctor will prescribe them to healthy genetic carriers. But experiments in genetic models will help to inform us about how effective these treatments might be in humans with genetic prion disease, and if successful, will provide additional evidence in support of moving to clinical trials.
why this drug?
Over the past several months we’ve been compiling a dataset of all the in vivo therapeutic trials in models of prion disease. It’s not quite complete yet, but we’re confident that at this point we’ve included all of the more successful therapeutic efforts in relevant models, most of which have their own blog posts.
Of all these experimental treatments, anle138b ranks as the most promising on a timepoint-delay plot. Here’s where it stacks up:
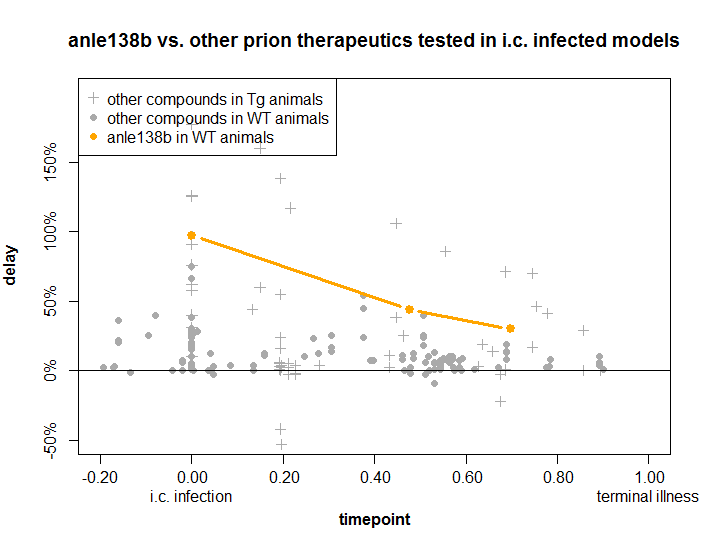
How to read this plot: the x axis is how long the researchers waited before starting the treatment. The later they wait (to the right), the harder it is to have an effective drug, but the more realistically the trial reflects what will happen with most real-life patients. The y axis is by what percentage the lifespan of the treated animals was increased as a result of treatment. Every point on this plot is a published in vivo experiment.
As you can see, a few treatments have been wildly successful in early stages, giving a 100% increase in lifespan, i.e. doubling survival. By the time you get 80% or 90% through the disease course, nothing works nearly so well. But of all the treatments that have been tested, the brand-new anle138b is the best. Even when administered about 70% of the way through the disease, it increased survival time by 30%.
That’s basically the best that has ever been accomplished. You will notice a few even more successful experiments denoted as gray marks above the orange anle138b line – but those were experiments in transgenic mice overexpressing PrP, which appear to have larger proportional responses to therapeutics than wild-type mice (and by implication humans) do.
Also, the compounds in those experiments are no longer really in the running as therapeutics. Pentosan polysulfate gave the strongest effects ever seen in animal models, but it can’t cross the blood-brain barrier and had to be infused directly into the brain. Clinical trials in the U.K. and Japan were unsuccessful [Bone 2008, Tsuboi 2009]. cpd-B also had large effects but proved highly strain-specific: it only worked on some rodent strains of prion and not others, casting doubt on its potential for human strains. 2-aminothiazoles are not included on this plot because the in vivo work is not published yet, but did have efficacy similar to that of anle138b. Unfortunately they proved ineffective against a human prion strain – sCJD prions tested in Tg(MHu2M) mice – and to our understanding the Prusiner lab is not pursuing further development on these compounds at this time.
anle138b has only ever been tested against RML prions in vivo, so it may yet prove to have the same strain specificity problems that have plagued 2-aminothiazoles and cpd-B. That’s one question we hope to contribute to answering in this therapeutic trial. There are some promising signs that anle138b’s effects might prove more universal than other compounds have. In vitro, anle138b appears to inhibit the oligomerization of several different misbehaved proteins including tau, alpha synuclein and huntingtin - and it also inhibits cell-free (PMCA) amplification of several different PrP prion strains [U.S. Patent US20110293520, Wagner 2013]. And that’s not all – in vivo, anle138b improved neuropathological and behavioral phenotypes in two mouse models of Parkinson’s disease (a rotenone model and an A30P transgenic model) [Wagner 2013]. Given that this compound appears to meaningfully interfere with oligomerization of entirely different proteins, we’re hopeful it will work well in different prion strains as well.
Dr. Armin Giese, the compound’s inventor, is ambitious and hopes to move anle138b to clinical trials within two years. Therefore we want to know how relevant this compound will be to human genetic prion diseases – and if the answer is “very”, then we hope that the data from our study will help to speed anle138b’s advancement through the regulatory process.
why these mice?
This study will use Tg(A116V) mice, which express a mouse PrP transgene with the equivalent of the human A117V mutation which causes Gerstmann-Straussler-Scheinker syndrome (GSS). These mice recapitulate certain key features of A117V prion disease in humans, including high plaque content, only modest spongiform vacuolation, low levels of insoluble PrPSc, and a long symptomatic phase (though the unique PrP cleavage pattern of GSS is only partially recapitulated) [Yang 2009]. In short, the model seems likely to be producing the same strain (conformation) of prion as real GSS does – or at least, something very close, increasing the odds that therapeutic results from these mice will translate well to humans.
While several other mouse models of genetic prion disease exist, Tg(A116V) is the only one that has been used in a successful therapeutic trial (of rapamycin) measuring survival time and time to symptoms [Cortes 2012 (ft)]. In any therapeutic trial, one needs to know the distribution of onset and survival times in order to do a power calculation, and one needs to know what phenotypes to look for as an endpoint for assessing therapeutic efficacy. These are known quantities for Tg(A116V) mice, and Dr. Mastrianni and his staff have experience from running the previous trial of rapamycin. In essence, our anle138b trial can be plugged into the existing system they have running, without needing to invent everything de novo. Moreover, untreated Tg(A116V) mice develop phenotypes at about 5 months of age, which makes a relatively rapid therapeutic trial possible, which means lower costs and quicker results.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.