Comparison of high-throughput screening approaches used for anti-prion small molecule discovery
This post will review high-throughput screening methods that have been utilized to discover anti-prion small molecules. First, here’s an at a glance overview of published studies I’ll cover in this post.
| study | format | target | compound library | number of compounds | prion strain | positive control | incubation time |
|---|---|---|---|---|---|---|---|
| Kocisko 2003 | ELISA | PrP-res | Spectrum Collection | 2,000 | RML & 22L | curcumin | 5 days |
| Bertsch 2005 | SIFT | PrPC / PrPSc binding | DIVERSet 1 | 10,000 | sCJD | DOSPA | N/A |
| Ghaemmaghami 2010 | ELISA | PrP-res | UCSF ChemDiv | 10,135 | RML | quinacrine | 6 days |
| Poncet-Montange 2011 | ELISA | PrP-res | Spectrum Collection | 2,160 | RML | quinacrine | 5 days |
| Leidel 2011 | ELISA | PrP-res | DIVERSet 1 | 10,000 | RML | suramin | 5 days |
| Karapetyan & Sferrazza 2013 | FRET | PrPC | US Drug Collection | 1,280 | N/A | tunicamycin | 1 day |
| Wagner 2013* | SIFT | PrPC / PrPSc binding | DIVERSet 2 | 10,000 | sCJD | DOSPA | N/A |
| Wagner 2013* | ELISA | PrP-res | DIVERSet 2 | 10,000 | RML | suramin | 5 days |
*Updated 2013-08-21. Wagner 2013 screened 10,000 DIVERSet 2 compounds using both the SIFT and PrP-res ELISA assays from Bertsch 2005 and Leidel 2011 respectively, and analyzed these in combination with the original DIVERSet 1 results from those studies.
And in the next three sections I’ll discuss the details of the three assay approaches used.
ELISA or “dot blot” assay for inhibitors of PrP-res formation
At its most basic, the idea here is to use PK digestion and and anti-PrP antibody to detect the existence of PK-resistant PrPSc. Variations on this concept has been used in low-throughput formats for a long time [Taraboulos 1990 (ft), Rudyk 2000 (ft), Winkhofer 2001 (ft)] and the was eventually adapted for high-throughput screening [Kocisko 2003]. In the high-throughput version, 96-well plates are filled with scrapie-infected mouse neuroblastoma (ScN2a) cells – about 20,000 cells per well – and incubated with various compounds for 5 or 6 days. Cells are then lysed, lysate is digested with proteinase K, and the lysate is transferred to a membrane that the proteins will stick to. Primary antibodies against PrP are introduced, then unbound antibodies are washed from the membrane, and fluorescently labeled secondary antibodies are bound to the original antibody. You get a fluorescent signal where PK-resistant PrP was present, and no signal where it was absent, which presumably occurs when a drug worked. Negative controls are simply untreated cells; positive controls require a compound known to interfere with PrP-res formation – people have used curcumin [Kocisko 2003], suramin [Leidel 2011] or quinacrine [Ghaemmaghami 2010, Poncet-Montange 2011].
SIFT assay for compounds that interfere with PrPC / PrPSc binding
SIFT stands for Scanning for Intensely Fluorescent Targets, but if you Google this you won’t find much. This is the name the authors used for their screening approach in one study [Bertsch 2005] and the concepts are more widely used even if the nomenclature is not. The goal of Bertsch’s SIFT assay is to detect compounds that interfere with PrPC binding to PrPSc. These two proteins are mixed in a very dilute solution, with PrPC labeled in green and PrPSc labeled in red. A laser is shone on the solution, and if red and green always wander through the laser’s beam a the same moment, that implies the two are bound – which is the default. If red often wanders through alone and green often wanders through alone, that means they’re unbound, and the test compound must therefore have inhibited their binding.
In practice, the PrPSc used was obtained from postmortem brain homogenate from sCJD patients, and the PrPC was recombinant mouse PrP 23-231. Bertsch never explains the motive for mixing mouse PrPC with human PrPSc – an odd choice, to my mind, given that the affinity between these two must be less than that of same-species PrPC and PRPSc. The recombinant mouse PrPC was labeled with Alexa 488 and human PrPSc was exposed to the L42 antibody which was in turn labeled with Alexa 476. (Interestingly, this implies that Alexa 488 binds to PrPC). DOSPA [Doh-Ura 2000] was used as a control, since this assay requires a compound that directly interferes with PrPC/PrPSc binding – compounds that act indirectly won’t do.
FRET assay for compounds that deplete PrPC
FRET shares with SIFT the conceptual similarity that two things are labeled in different colors and the assay detects whether the two things are in proximity to each other or not. In FRET, two different antibodies are labeled with different excitatory wavelengths – say, red and blue. A red laser excites the red antibody and – if and only if the two antibodies are in close enough proximity for resonance to operate – a blue wavelength will be emitted by the other antibody. FRET assays are often used to check whether two things have come into (or left) proximity with one another as a result of a compound treatment. In that sense, it’s conceptually similar to SIFT (above).
This particular assay at Scripps Florida [Karapetyan & Sferrazza 2013], though, used antibodies against two different epitopes on PrP. If PrP is present, the two antibodies will bind to the same PrP molecule, acquiring enough proximity to resonate. If PrP is absent, the antibodies will just float around aimlessly and not necessarily be close to one another. In this way, the FRET assay is used to quantify PrP and thus to screen for compounds that deplete PrPC.
In practice, the assay was applied to whole cells, not lysed or pelleted, meaning that it only detected PrPC on the cell surface and in the culture medium (such as shed PrP). The authors cited the fact that using PIPLC to cleave PrP’s GPI anchor, releasing PrP from the cell surface, can cure ScN2a cells of prion infection [Enari 2001], as evidence that cell surface localization is crucial for prion infectivity. In fact, the relationship of PrP shedding – the closest in vivo analogue to PIPLC treatment – to prion disease is more complicated. However, since the culture medium was not washed away in the Scripps study, the assay would not actually have detected compounds that increased PrP shedding anyway. The assay also would not have detected compounds that reduced intracellular PrP. Intracellular PrP could well matter in prion disease – for instance, cytosolic PrP has been implicated in proteasome dysfunction [Yedidia 2001, Ma & Lindquist 2001, Kristiansen 2007 (ft)], and so it would be nice to detect compounds that reduce intracellular PrP as well. However it doesn’t seem too likely that there are many compounds out there which could reduce intracellular PrP without affecting cell surface PrP, so this is probably not a major problem with the assay.
Of note, the two antibodies used for FRET in this assay were SAF32, which targets the octarepeat region, and D18, which targets amino acids 133-157 (alpha helix 1). These epitopes flank the central region of PrP, and therefore both alpha and beta cleavage would have abolished the proximity of the two antibodies, and thus compounds that promote either of these events would have turned up as assay hits.
which mechanisms of action does each assay approach afford us the opportunity to discover?
Consider the life cycle of PrP in an infected cell as diagrammed below.
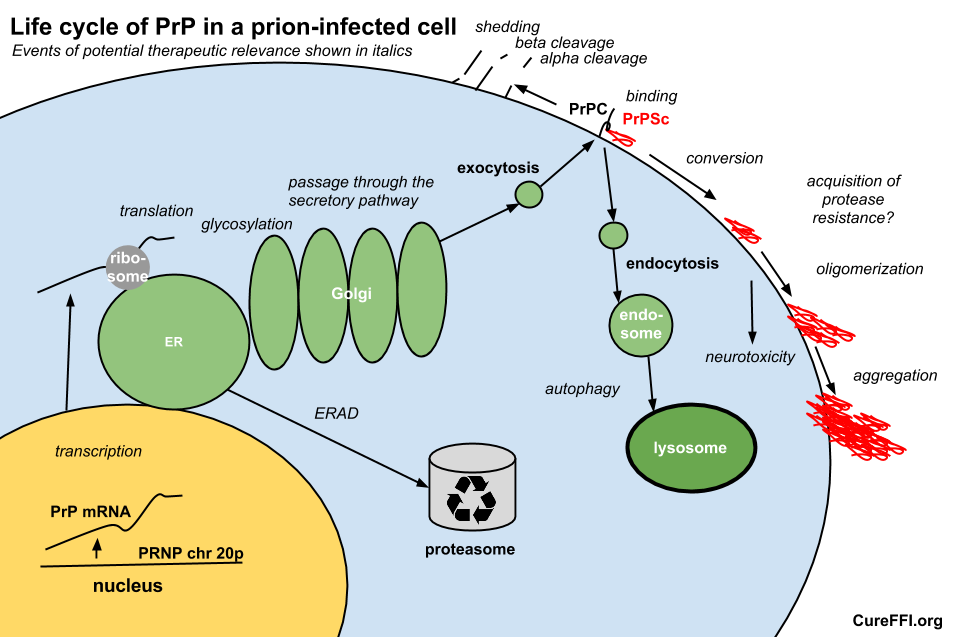
Therapeutically relevant events in PrP’s life cycle could include transcription, translation, passage through the secretory pathway including glycosylation, enzymatic cleavage, shedding, degradation via lysosomes and proteasomes, binding to PrPSc, conversion to PrPSc, oligomerization, conformational change including acquisition of protease resistance, and aggregation.
Of these events, SIFT is exclusively designed to detect compounds that interfere with the binding event. The Scripps FRET assay can only detect events in PrPC’s life and nothing from the PrPSc binding onward. Advocates of the PrP-res ELISA assay will point out that it can detect compounds with a wide variety of mechanisms of action. Indeed, one such study has suggested a scarcity of compounds that directly bind to PrP [Poncet-Montange 2011], and only a handful of hits affected PrPC levels, which may be taken to caution against devising assays that are too specific in their focus.
Of course, the PrP-res assay is not universal either. Since scrapie infection isn’t toxic to N2a cells, there’s no way to identify compounds that lower the neurotoxicity of PrPSc without reducing its quantity. And like any assay, it’s by necessity a highly simplified in vitro model. There are are plenty of things that could be relevant in vivo – say, the still-mysterious role of glial proliferation in prion disease – that won’t come through.
what to do about strain specificity?
Perhaps a bigger problem is that so many of the compounds that fare very well as PrP-res inhibitors – quinacrine, cpd-B, curcumin, 2-aminothiazoles – prove to be highly strain-specific in their efficacy. The major advantage of an assay directed at depleting PrPC, such as FRET, is that even if it’s harder to find hits, at least you know that the hits you do find are pretty likely to act universally across strains of prions. That’s hugely important – both because human prions come in several different varieties and because human prions still can’t be propagated in cell culture, forcing our reliance on rodent cell models for screening.
There are also several ways to accommodate a search for strain insensitive drugs within the PrP-res paradigm. One approach is to do primary screening in cells infected with one prion strain and then counterscreen with a different prion strain – say, RML and then 22L [Kocisko 2003]. If only we had human cell models of prion propagation, one or both of the strains tested could be human strains. A different approach is to screen for inhibitors of the new drug-resistant strains that come out of chronic treatments with the same compound – Kurt Giles has suggested this with regards to drug-resistant prions created by IND24 treatment.
Another concern with assays directed at PrP-res is how well the quantification of PrP-res tracks with total PrPSc. In RML, the strain almost always used in these assays, most PrPSc is protease resistant [citation needed]. But in sCJD, only 10% of PrPSc is protease resistant [Safar 2005]. In fatal familial insomnia there is very little PrP-res [Medori 1992 (ft), Brown 1995, Jackson 2009], and in VPSPr there is zero [Zou 2010]. It is an open question how well inhibition of PrP-res formation in this assay predicts inhibition of total PrPSc accumulation.
Assaying for protease-sensitive PrPSc would be more of a pain than PrP-res, involving quantifications of antibody binding both before and after denaturation. That’s harder to make high-throughput. One alternative would be assaying for total PrP in infected cell lines, which would be no extra hassle, indeed, slightly easier than assaying for PrP-res. I would like to see a breakdown of what fraction of total PrP in ScN2a cells is PrPC, sPrPSc and rPrPSc (aka PrP-res) – this has been measured (though not presented as such) for many strains in vivo [Safar 1998] but I can’t find a reference for these quantities in RML-infected N2a cells, which are the model used in high-throughput assays. Presumably, assaying for total PrP would somewhat decrease power to detect specific inhibitors of rPrPSc formation, while gaining (any) power to detect inhibitors of sPrPSc and (importantly) enriching hits for compounds that affect both sPrPSc and rPrPSc. It would also maintain approximately the existing power to detect compounds that reduce PrPC altogether.
why aren’t PrPSc-promoting compounds reported?
None of the above studies reported on any compounds that increased PrP-res formation. Perhaps that’s because no one is going to waste their time validating such undesired compounds in counterscreens or animal models, and no one wants to report unvalidated results. Still, it would be quite valuable to know the identity of any such compounds. They might give us hints to some underlying biology. And for approved drugs, they might merit recognition as potential environmental risk factors for sCJD or potential drugs for genetic mutation carriers to avoid. Some of the same drugs have come up more than once as PrP-res inhibitors (e.g. astemizole [Kocisko 2003, Karapetyan & Sferrazza 2013]) so if the same is true of compounds that increase PrP-res, then the studies would provide some validation of one another even if no one validated the results individually.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.