2-aminothiazoles: a tale of drug resistance and strain specificity
background
One of the biggest discoveries to emerge from the past few years of prion research is that prions are capable of evolving and adapting.
Some of the earliest convincing evidence for this phenomenon came from attempts to figure out why quinacrine didn’t extend survival in animal models (or human patients) despite being miraculously effective at clearing PK-resistant PrPSc in cell culture [Ghaemmaghami & Ahn 2009]. Interestingly, quinacrine did seem to very slightly extend survival in some RML prion-infected mice, but only with a brief treatment regimen and not under continuous treatment [Table 1]. It turned out that the drug caused only a transient (<20 days) reduction in PrPSc in vivo. In cell culture, quinacrine appeared to work for at least a week as long as cells were kept continuously dividing. But once cell division was arrested, the drug worked for only about 2 days. There was one good explanation for all these phenomena: quinacrine was selecting for quinacrine-resistant prion strains. This conclusion was also supported by the biochemical characterization of the new resistant strain: it lost its PK resistance at lower GdnHCl concentrations than the native RML strain did [Table 2]. (CDI also indicated different conformational stability for the resistant prions).
Work published the following year extended this finding to the glycosylation-modifying drug swainsonine, and showed that prions in cell culture can evolve to optimize for cell culture propagation while prions in vivo can evolve to optimize for in vivo propagation [Li 2010]. That study also employed a cell panel assay – testing which prions can infect which cell lines – to characterize the properties of newly evolved strains.
There’s been a great deal of work in this area in the past few years, and drug resistance was among the major topics at Prion2013. A new review [Miller-Vedam & Ghaemmaghami 2013] chronicles the discoveries and discusses their implications. While we still don’t understand exactly what this strain evolution entails at the molecular level, all evidence seems consistent with each “strain” (e.g. RML, ME7, MM1 sCJD) being a collection of substrains, each of which represents a particular conformation of PrP. Drug treatments may target only one substrain, meaning they select for the others and also meaning they may be ineffective against other strains that aren’t enriched for the particular substrain targeted.
How this one protein can adopt such an apparently huge number of distinct self-propagating and disease-causing conformations remains a mystery. But an excellent new paper from David Berry and Kurt Giles at UCSF shows clearly the implications for drug discovery [Berry 2013]. I first blogged about this work after seeing their poster at Prion2013, but the new paper is so dense with data and analysis that this post will give it a second, more detailed look.
2-aminothiazoles are really effective – against some strains
IND24 (above) and IND81, the two flagship compounds from this new class of drug candidates, the 2-aminothiazoles (background here), are really effective against RML prions, as well as ME7 and CWD prions. Along with cpd-B and anle138b, these drugs form a third class of highly effective anti-prion small molecule. All three have comparable effects in vivo, as shown on this timepoint-delay plot:
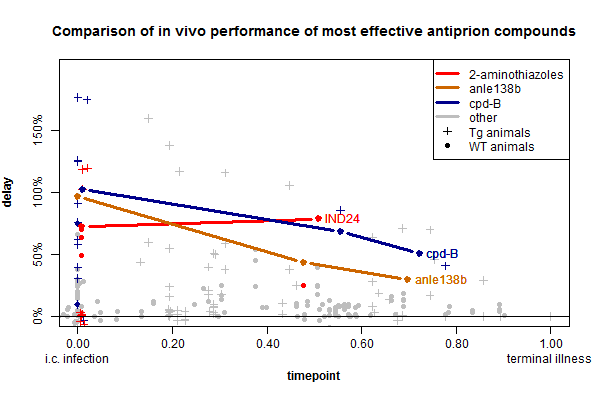
(Data from my ongoing prion therapeutic review, and here is the R code used to create this plot).
I’ve color-coded all of the reported experiments involving anle138b, cpd-B or 2-aminothiazoles. Additionally the thick lines represent the timepoint-delay relationship for treating RML prion infection in wild-type mice according to [Wagner 2013, Lu & Giles 2013, Berry 2013]. I chose to highlight these experiments because they represent the most standard model used for in vivo prion research and are the most directly comparable between the three compounds.
the surprising lessons of this study
In the particular model highlighted above, the 2-aminothiazole IND24 has efficacy on par with the other compounds and surprisingly, its efficacy appears not to diminish much at timepoint 0.50 compared to timepoint 0. When mice were treated at 1 dpi, their lifespan was extended by 73%, and when they were treated at 60 dpi (timepoint ~.50) it was extended by 79%. We would need more timepoints to say how broadly this is true, but IND24 does appear to be at least somewhat unique among the antiprion compounds discovered so far in that its efficacy diminishes little with late administration up to the halfway point. Up until now, it had appeared that the negative slope of timepoint-delay curves was an inescapable limitation of prion disease treatments, common to cpd-B and anle138b as well as pentosan polysulfate, amphotericin b and other treatments. The suggestion that some treatments might not lose much efficacy with time is at least potentially encouraging news.
The other points on the plot which I have color-coded but not drawn lines through represent other experiments with the same drugs, against different prion strains or in different mouse models. IND24 was tested in a battery of conditions, with data reported in Tables 1 & 2 of [Berry 2013]. You’ll notice a barrage of red data points in the lower left corner of the plot above – these represent experiments with IND24 where it wasn’t so effective. These experiments fall into two categories: drug resistance and strain specificity.
The authors present an impressive array of evidence in Fig 2 to show that the properties of the RML prion strain changed when mice were treated with IND24. Its conformational stability changed (measured by how high of GdnHCl concentrations it can withstand while remaining PK resistant), its glycoform ratio changed, its neurotropism (preference for certain brain regions) changed, its incubation time (in untreated mice) changed, and above all, its sensitivity to further treatment with IND24 was greatly diminished. All of this argues that the strain had evolved resistance to IND24.
Interestingly, the resistant strains still seemed to “remember” their origins – perhaps the dominant conformations within native RML were still present at low quantities in the resistant strain, or were an easy mutational conformational change away from the resistant conformation. In any case, when the strain was passaged again in mice without IND24, it appeared to gradually revert to original RML.
Even more disturbingly, cell culture experiments showed that the IND24-resistant prions were also resistant to cpd-B and quinacrine. This suggests that these three anti-prion compounds may all act on the same or a similar subset of prion conformations. This leaves open the question of whether we’ve yet discovered any anti-prion small molecules that don’t share this property.
Just as IND24 didn’t kill off all the conformations of prion found in RML, it appears not to have killed off any of the conformations found in the human MM1 or VV2 sCJD prion strains. IND24 was completely ineffective in vivo against both of these strains. For the record, the strain specificity of IND24 can be conclusively tied to conformation and not PrP amino acid sequence, because in the same chimeric mouse model, Tg1014 [Giles 2010], IND24 was effective against RML and not against MM1 sCJD. (The same argument can be made for cpd-B by comparing RML to Fukuoka-1 and 22L data in [Kawasaki 2007]).
When I first saw that data, the phenomenon that came to mind was regression to the mean. If prion strains are conformationally different from each other, then an inhibitory compound will have a different effect magnitude on each of them. By discovering anti-prion compounds in screens based on the RML strain, we select for the compounds most effective against RML, while we have not similarly selected for compounds most effective against other strains. This means that on expectation, the compounds we discover will be less effective against other strains than they are against RML.
While this may be true and I stand by the basic reasoning above, the new paper also presents encouraging evidence that anti-prion compounds can have serendipitous efficacy against prion strains that they were never screened against during development. Although IND24 was ineffective against the two human CJD strains, it was effective in vivo against ME7 and CWD. This is especially surprising for ME7 because IND24 actually had been tested against ME7 in cell culture and did not look very effective.
implications for anle138b and future drug discovery efforts
The strain specificity issue demonstrates that compounds effective against RML won’t necessarily work for human prion strains. So far, anle138b has only been tested in vivo against RML. This is one motivation for our current efforts to test anle138b against models of the A117V GSS, FFI and E200K strains.
Of course, anle138b was tested in vitro using PMCA against vCJD and multiple sCJD subtypes, and it appeared equally effective against all strains tested [Wagner 2013]. As of now, there’s not yet any data to validate that a compound’s inhibition of PMCA reactions is predictive of its inhibition of prion replication in vivo. If we could demonstrate that inhibition in PMCA (or any other in vitro assay – say, RT-QuIC) is indeed predictive of a compound’s strain specificity, then we could shave years off of drug development.
As I understand it, the current thinking on the drug development pipeline in Prusiner’s group at UCSF is roughly as follows:
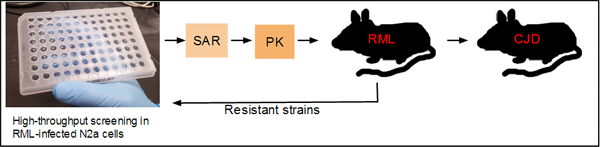
Compounds discovered in screening must first go through extensive structure-activity relationship (SAR) and lead optimization studies and muster decent pharmacokinetics before they ever see a survival study in an RML prion-infected mouse model. Only those compounds that are effective in RML move on to mouse models infected with human prion strains. To address the drug resistance issue and discover drugs that kill resistant strains, the resistant strains can be transferred to cell culture and screened against in another high-throughput assay.
Such a pipeline is an impressive achievement. Throughout the 1990s and early 2000s, anti-prion drug development was completely ad hoc, with candidate compounds chosen based on intuition or serendipity. To have multiple high-throughput assays, chemical biology and medicinal chemistry teams capable of optimizing the screening hits, and then a series of reasonably fast mouse models for assessing in vivo efficacy, including against human strains, is a tremendous improvement over where the field was a decade ago.
Still, it’s a shame that compounds have to go through years of development before ever seeing a human prion strain for the first time. I’m not sure when the actual research occurred, but judging by publication dates, 2-aminothiazoles didn’t make it to testing against CJD prions until ~3 years after their discovery [Ghaemmaghami 2010, Berry 2013].
One solution to this problem would be the development of human cell lines capable of propagating prions, a challenge which many bright people have attempted and failed [mostly unpublished rumors]. But even if we had, say, an MM1 sCJD-infected cell line, one still wonders whether that line would have any predictive power for VV2 sCJD or FFI or the several other prion strains found in human patients.
Another is to only pursue approaches that (we think) will be non-strain-specific. After doing his own work on prion strain evolution, Charles Weissmann concluded that it was best to abandon screening in infected cells altogether and go after PrPC as the drug target [Li 2010], an approach which he and Corinne Lasmezas are now pursuing at Scripps Florida [Karapetyan & Sferrazza 2013].
Yet a third possibility, and the one I find most intriguing, would be the development of an in vitro assay that predicts strain specificity. Such an assay could be quick (compared to bioassays), could be done in parallel on several strains of interest, wouldn’t require favorable pharmacokinetics and so could sit near the beginning of a pipeline such as the one diagrammed above. This would only work for compounds that directly interact with PrP, but it would still be of great value. Does the fact that anle138b inhibits sCJD in PMCA really mean it will be effective against sCJD in vivo? If the answer is yes, and if this can be validated for a few different compounds in parallel in vitro and in vivo experiments, then the implications for anti-prion drug discovery would be huge. An assay that filters, or even slightly enriches, for screening hits that look universal in their efficacy could shave years off of the drug development timeline.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.