What can loss-of-function variants predict about drug safety?
This post (and much more) is now a bioRxiv pre-print: [Minikel 2019]. See this post for more information!
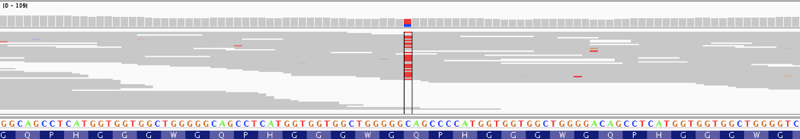
Sequencing reads showing a nonsense variant in PRNP, a gene for which antisense oligonucleotides to lower expression are in development.
introduction
Human genetics can tell us a lot about how to design better drugs. From beautiful narratives of how individual drugs were (or could have been) informed by human genetics [Plenge 2013] to statistical analyses showing that drugs whose targets are validated by human genetics are more likely to reach approval [Nelson 2015], there are plenty of reasons to believe that drug target selection should focus on genes known to be causal in disease.
Some of this body of thought focuses on drug efficacy, specifically the idea that: 1) many/most drugs that fail in clinical trials fail on lack of efficacy, 2) many/most failures for lack of efficacy are because the drug’s target is not actually causal for the disease in question, and 3) the number of failures can therefore be reduced by choosing targets whose genetic evidence for causality is unimpeachable.
The question I want to talk about in this post is how genetics can inform on drug safety. My specific question is whether loss-of-function (LoF) variants can be a model for lifelong inhibition of a drug target, and therefore can inform on whether or not a drug would be expected to suffer from on-target toxicity. This was the subject of an analysis I led in the MacArthur lab around 2015 and which we never wrote up. To this day I find that this question has a way of coming up in conversation or in Q&A at talks every few months, so I decided it was time for a long-delayed blog post.
Long story short: there is no doubt that if you’re developing a drug against gene X, then knowing the effect of human loss-of-function variants in gene X is incredibly valuable. Yet at the same time, there is no simple formula or algorithm for what pattern of LoF variants makes a gene a safe target or an unsafe target. In fact, there isn’t even any complex formula or algorithm. The best we can do is deep curation and consideration to each individual gene and drug.
premise & hypothesis
Variants annotated as nonsense, frameshift, or essential splice are categorized as protein-truncating variants (PTVs). These in turn are generally predicted to cause loss-of-function (LoF), though of course there are important exceptions. In this post I’ll use the term LoF rather than PTV.
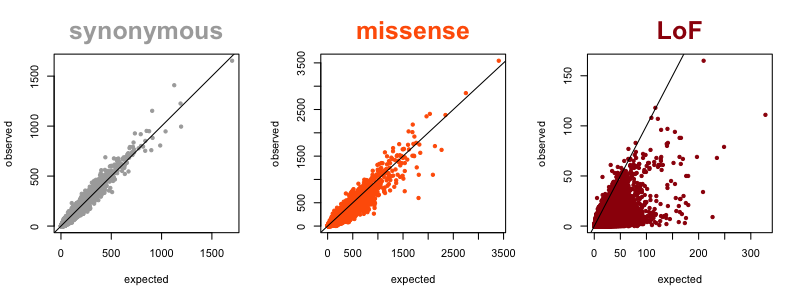
Now that we have large reference datasets like ExAC/gnomAD, we can quantify the degree of natural selection against LoF in each gene by looking at how many such variants are observed in a gene, compared to prediction based on mutation rates. A gene that is heavily depleted for such variants compared to expectation is said to be constrained. My colleague Kaitlin Samocha worked out the concept and math for all this in [Samocha 2014] and further refined it in section 4 of the supplement of the ExAC paper [Lek 2016]. In the figure below, each dot represents one gene, and its position on the plot represents the expected (x axis) and observed (y axis) number of variants in ExAC v1. For synonymous variants (where we expect minimal natural selection), the correlation is excellent, with almost all dots lining up right on the diagonal. For missense variants, you see increased density below the diagonal, because some genes are very intolerant of missense variation. For LoF variants, you see most of the density below the diagonal, because most genes are at least somewhat intolerant of LoF variation, and some genes extremely so.
Several different metrics have been developed to quantify the degree of constraint on individual genes [Fuller 2018]. For this post, I’ll simply use the metric obs/exp, the ratio of observed to expected LoF variation, to quantify intolerance to loss-of-function.
The hypothesis, then, is:
If a gene is severely depleted for LoF variation, then a drug candidate that targets that gene is more likely to cause adverse events.
proof that this problem is non-trivial
It’s easy to use a few examples to convince yourself that any sweeping, absolute generalization will be false. For instance, if you imagine that genes that are completely depleted for LoF variation cannot possibly be viable drug targets, you’d be wrong, because MAOA and MAOB each contain ~0% of the expected amount of LoF variation in ExAC, yet have long been successfully targeted by approved monoamine oxidase inhibitors (MAOi) such as selegiline. (Relatedly, there are also examples to show that a lethal mouse knockout phenotype doesn’t mean a gene can’t be a drug target — for example, statins target HMGCR, but constitutive or liver-specific knockout of its mouse homolog are lethal [Ohashi 2003, Nagashima 2018].) In any case, at best, we’re going to find a correlation, not a hard-and-fast rule.
But even drawing correlations is going to be difficult, because many genes are the targets of both safe, approved drugs and of unsafe or withdrawn drugs. PTGS2 (also known as cyclooxygenase 2 or COX-2), which is completely depleted for LoF variation (0 observed, 20 expected) is the target both of rofecoxib (notoriously withdrawn) and celecoxib (still widely prescribed) — as well as, you know, aspirin. HTR2B (serotonin receptor 2B), which has 100% of its expected LoF variation (13 observed, 13 expected), is the target of a whole swathe of molecules: olanzapine (an antagonist), dihydroergotamine (an agonist), methysergide (a no-longer-recommended antagonist), fenfluramine (a withdrawn agonist), and MDMA (an illicit street drug).
And on reflection, there are obvious reasons why this should be non-trivial. Genetic knockout is lifelong while pharmacological inhibition can be temporary; genetic knockout has a specific “dose” (perhaps 0% of wild-type gene function for homozygous LoF and 50% for heterozygous LoF in the simplest case) while the dosing of pharmacological inhibition can be titrated as needed. In addition, there are myriad ways in which chemical perturbation and genetic knockout can have divergent effects. A drug might target a complex, or both of two paralogs, or only one functional domain of a protein, and so on. Stuart Schreiber gives a nice list of such examples in his Chemical Biology class.
quick-and-dirty analysis
Above caveats having been made, what does the trend look like? For instance, how does the amount of LoF variation in the genes targeted by approved drugs — our success stories — compare to other genes?
Here is the mean (and 95%CI indicating our certainty about that mean) obs/exp value for drug targets (N=286, from DrugBank via the MacArthur Lab gene_lists repo) compared to all genes, as well as a number of other gene lists for context:
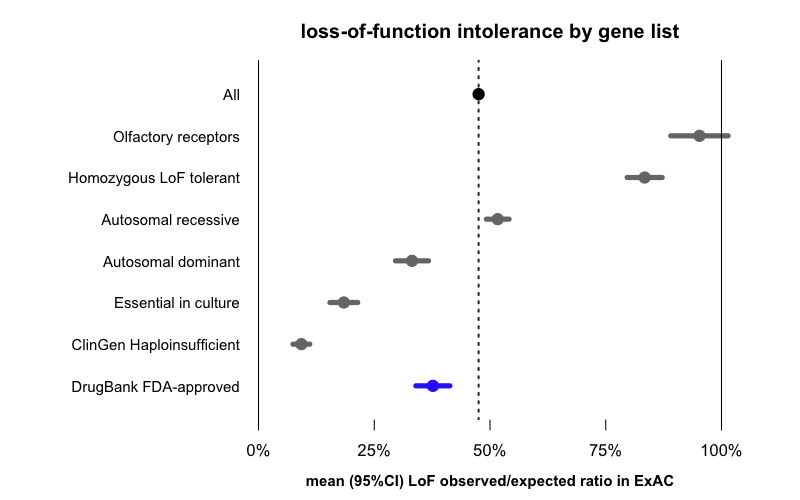
The average gene in general has 48% of its expected amount of LoF variation. The numbers for the other gene lists fall out exactly as you’d expect: olfactory receptors, which are notoriously dispensable in humans, have almost 100% of their expected LoF variation, and genes known to tolerate homozygous LoF in humans also have more LoF variants than the average gene. Recessive disease genes are close to average, while dominant disease genes are more depleted for LoF, and genes known to be essential in cell culture or associated with diseases of haploinsufficiency are even more severely depleted.
Against this backdrop, what may seem surprising is that drug targets are actually on average more depleted for LoF than all genes are. But is this really surprising?
problem 1: confounding by gene class
When drugs are broken down by target class, most of them fall into a few large families of classically “druggable” targets [Russ & Lampel 2005]. We looked at the classically “druggable” families listed in the Guide to Pharmacology, as well as a list of kinases from UniProt. In our dataset, these two lists collectively account for 65% of drug targets. If we look at the sets of all of these genes (regardless of whether targeted by drugs), we find that these too are depleted for LoF variation compared to all genes.
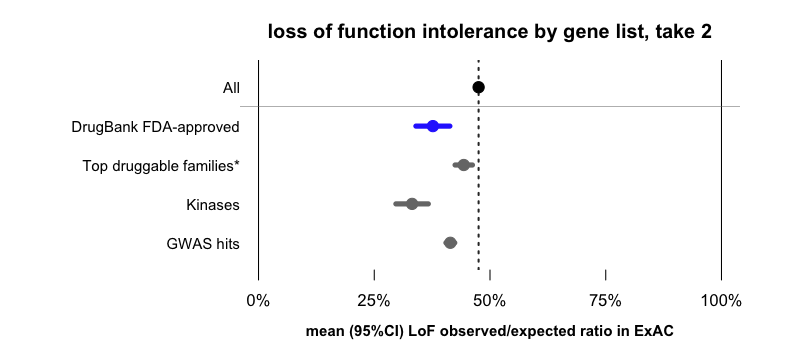
*Erratum: a previous version of this post incorrectly labeled this data point as “GPCRs”. In fact, it includes GPCRs as well as ion channels, catalytic receptors, transporters, and enzymes catalogued by Guide to Pharmacology.
One story you could tell here is that it’s not necessarily the case that more strongly LoF-constrained genes make better drug targets. It’s just that, at least as of this historical moment, the pharmaceutical industry has so far been relatively successful at finding chemical matter against certain families of proteins that happen to be more LoF-constrained than average.
That is just one example. I’ll briefly mention another, also shown in the above figure. At least some drug targets were selected because of their association to human disease, and it’s also been shown that genes that contain GWAS hits (i.e. disease-associated genes) are on average more LoF-constrained than other genes — see Fig 3E and Supp Figure 5 of [Lek 2016]. In the dataset used here, drug targets are about 2.4-fold enriched for GWAS hits (P = 2 × 10-10, two-sided Fisher’s exact test), and GWAS hits, like GPCRs and kinases, are more depleted for LoF variation than the average gene.
The set of drug targets is not some elegant, random list that fell from the sky and is magically orthogonal to all other lists. It’s incredibly prosaic and deeply influenced by all kinds of variables we can observe (protein class and “targetability”, involvement in human disease, historical accident) and probably plenty of variables we cannot observe. So there is plenty of opportunity for a Simpson’s Paradox here, where maybe drug targets are more LoF-constrained than the average gene, but may be less LoF-constrained than average after controlling for X, but are more LoF-constrained again when you also control for Y, and so on. But we just don’t have the N needed to endlessly stratify and resolve a Simpson’s Paradox here, because there are only on the order of a thousand drugs, and only on the order of hundreds of genes known to be targeted by them.
problem 2: lack of the right annotations
In the forest plot above, what I was showing was the average degree of LoF depletion in each gene class. While this enables an easy comparison, it hides the full distribution, which is arguably equally or more informative.
Here’s the distribution of LoF obs/exp values for drug targets and for all genes:
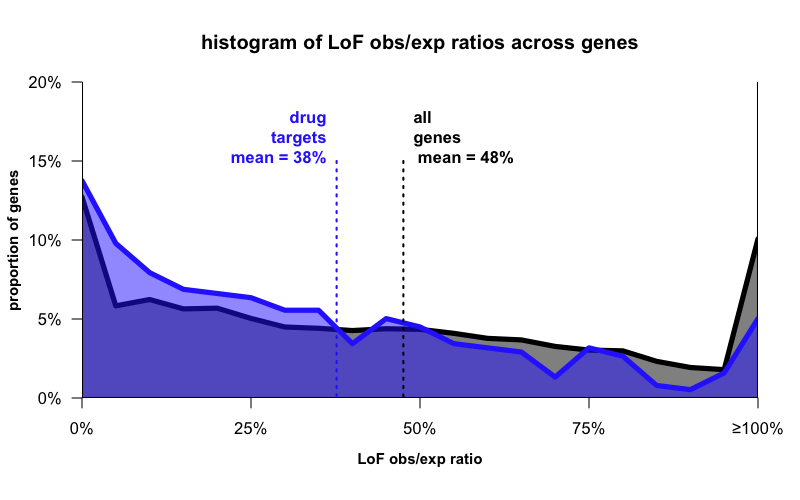
What you see now is that while the means differ a bit, the distributions aren’t so different. Drug targets, like all genes, are all over the map — some are not at all depleted, and some are completely depleted, for LoF variation. As I said earlier — a single hard-and-fast rule won’t work here.
But is that surprising? After all, we haven’t even filtered for which drugs even act negatively on their target’s function (antagonists, inhibitors, etc), and there exist plenty of drugs that increase their target’s function or cause some neomorphic gain-of-function. We also haven’t asked which drugs are taken chronically (a case where genetic knockout might more closely mimic the drug’s effect) as opposed to something you take for a week, or something you apply topically in a local area. We haven’t filtered for how severe the condition is (worse adverse events being able to fit within a favorable risk-benefit balance if the condition is more severe). And perhaps worst of all, we’re just comparing targets of approved drugs (successes) to all genes (neutral), whereas we’d expect to get more signal if we had a negative set to compare to — drugs that failed for on-target reasons.
Earlier I said the hypothesis was:
If a gene is severely depleted for LoF variation, then a drug candidate that targets that gene is more likely to cause adverse events.
But if we could rephrase this the way we’d really like to ask the question, it would be:
The targets of inhibitors intended for chronic use in non-fatal conditions which fail early in development due to severe on-target toxicity are particularly depleted for LoF variation.
But unfortunately, that’s just not a question we have the data to ask. Each bolded word in the above statement represents something where we just couldn’t find an appropriate dataset:
- inhibitors — It is known how many drugs affect their targets, and I would bet that among drugs where the target is known at all, the effect on the target (e.g. inhibitor, activator, etc) is almost always known. But we couldn’t lay our hands on a clean, well-annotated list that we trusted. It could be done with a lot of manual curation. But even then, as noted above, some genes are targets of both inhibitors and activators, so things get noisy.
- chronic use — Prescribing information is publicly available from Drugbank and FDA databases and so on, it’s just not trivial to extract a short enumerated list in order to do analysis.
- non-fatal conditions — Again, indications for drugs are publicly available, it’s just not trivial to match up indications to severity in an elegant, analysis-ready way.
- fail early in development — Here we get to the really hard part. We know what drugs get approved, which are our success stories. We know what drugs get withdrawn, which might seem like failures but really, these are the very-very-close-to-success stories, so we don’t expect to see much signal comparing those to the drugs that are still approved. We have some data on what drug candidates get to clinical trials, but really, these too are the not-so-very-far-from-success-either stories, because they at least made it through all the stages of preclinical development and often Phase I. What you really want to know about are things that failed very early in the pipeline, and that’s not information that drug companies are usually keen to give out.
- on-target. This is the biggest problem here. When a drug does fail in preclinical development or in Phase I, it’s often unclear to the public’s eye whether it was because of safety or because of efficacy or because of “business reasons”. And even if the answer is “safety”, it is very difficult to figure out the molecular source of adverse effects. Moreover, once a drug has failed there is little incentive to figure out why it failed. It makes more sense for the company to just move on to the next thing. I am guessing that even pharma companies, in most cases, could not confidently tell you which of their failed drugs failed due to on-target toxicity.
solution: deep curation
All the above points make it clear that even if LoF variants are incredibly informative for drug discovery, and I think they are, we’re never going to have a plug-and-play formula. We are never going to be able to say, for every X% your gene is depleted for LoF, your probability of adverse events goes up by Y%. And we are certainly never going to have a rule, like don’t develop drugs against targets with >Z% depletion of LoF variation.
But that’s okay, because remember, developing a drug is a monumental investment. It is an undertaking so large, and requiring such an intimate understanding of the drug and the target and the indication and the patient population and so on, that no one was going to be swayed by a correlation or a forest plot anyway.
What can we do instead? A lot, we think.
A first step in any analysis is to curate whether the protein-truncating variants you are looking at in a gene are really LoF or not. Konrad Karczewski’s LoFTEE has done a great job with throwing out the obvious error modes (rescued splice sites, very late nonsense that don’t trigger nonsense-mediated decay and so on), but there are still loads of variants that may look like LoF and not be. C-terminal PTVs in PRNP are actually gain-of-function [Minikel 2016]. The PTVs in ASXL1 in ExAC are actually somatic variants [Carlston 2017]. PTVs might be in an exon not expressed in your tissue of interest, or not expressed in the most essential version of the transcript. In general, inspecting the positional distribution of PTVs observed in ExAC (and in disease cohorts) is critical — if they are clumped together and not scattered randomly throughout the gene, there is usually a reason.
If there is a known disease association for your gene, you want to be very certain of whether the disease mechanism is loss-of-function or gain-of-function. Even in very well-studied genes where the answer perhaps should be obvious, one can still find people who debate this issue. For instance, when reading up on the disease mechanism of SOD1 mutations for my term paper for protein-folding class I was amazed to see that one can find papers claiming SOD1 causes ALS by a loss-of-function, even though the evidence for a gain-of-function disease mechanism appears overwhelming. You’ll never convince everyone, but you at least want to convince yourself that you’re confident of the disease mechanism. Examining the proportion of missense vs. protein-truncating variants annotated as pathogenic in ClinVar and other databases can be valuable, as can examining the positional distribution of reportedly pathogenic variants. One elegant example of this is in [Shaw & Brand 2017] — inspect Figure 3 and notice how the positional and functional (missense vs. PTV) distribution of variants in SMCHD1 differs between those that cause arhinia (a gain-of-function syndrome), FSHD2 (a loss-of-function syndrome), and variants seen in ExAC controls. Another example is the PRNP work mentioned above [Minikel 2016].
Once you’ve worked through whether the PTVs in question are truly LoF, and whether LoF causes any known disease, you can look at aggregate statistics about LoF variants in your gene of interest to gain some insights. In some cases, especially for longer genes where the expected number of LoF variants is high a priori, the cumulative allele frequency of LoF variants may be enough to tell you something powerful. If, say, 0.1% of people have a heterozygous LoF variant in your gene of interest, and yet no disease is known to be associated with haploinsufficiency of that gene, then heterozygous LoF is probably at least reasonably well-tolerated, since a genetic disease with a prevalence of 1 in 1,000 people would have been discovered by now if it was even remotely severe and modestly penetrant. Of course, that doesn’t rule out some fitness effect of LoF, so you also want to look at evidence for depletion of LoF variants overall, say, using the obs/exp metric employed here.
Then comes the type of deep curation effort for which the capability is only now emerging. Where you can gain access to limited individual-level data, you want to look at whether the LoF individuals are enriched in any one cohort. For instance, SETD1A LoF variants are found in ExAC, but are mostly in ExAC’s psychiatric cohorts, and do turn out to show convincing association to psychiatric disease [Singh 2016]. Where age data are accessible, you also want to know whether the age distribution of these individuals skews young, consistent with predisposition to some late-onset disease. And of course, the ultimate dream is to have access to rich individual-level phenotype data and ability to recontact people. We’ve been able to do this on a very limited basis for a few genes through ExAC cohorts — PRNP, CIRH1A [Minikel 2016, Lek 2016], but with new cohorts like NIH’s All Of Us being designed to enable these types of studies, hopefully this capability will become more common in the future.
Once you have all this info, what do you do with it? You consider it in the context of the specific drug you’re developing and the specific indication you’re developing it for. The data might increase or decrease your confidence that there exists a therapeutic window, or suggest additional experiments to do. Because even totally LoF-depleted genes can be viable drug targets, it’s probably a rare case that the data will talk you out of developing the drug altogether. But the evidence from human genetics might provide insights on the level of inhibition or knockdown that one should aim for, or the types of adverse events one should be most on the lookout for, or the patient population you want to go into first. Nothing about developing a drug is trivial, and that includes applying lessons from human genetics. But the fact that it’s non-trivial shouldn’t stop us from reading out through human genetics the incredibly valuable data from experiments that nature has already done.
why I’m writing about this now
As I said up top, this analysis from 2015 has a way of continuing to come up on conversation today, even after I’ve shifted gears away from human genomics and towards drug and biomarker discovery. Whenever Sonia and I give a talk about our goal of lowering brain prion protein (PrP) with an antisense oligonucleotide, an astute audience member will always ask, what is the PrP knockout phenotype? And by implication, will it be safe for a drug to lower PrP?
I gave a quick answer to this question here, but in truth there are two levels on which one can answer such a question.
The first question is, what is the evidence that a reduction in PrP gene dosage is okay? Here we have just about the cleanest bill of health you could ask for. In mice, the homozygous knockout phenotype is mild and no heterozygous knockout phenotype is known [Bremer 2010]. In humans, there is no evidence for constraint against loss-of-function mutations, and the loss-of-function heterozygotes we’ve been able to identify are healthy [Minikel 2016].
The second question is, even if there were a more severe knockout phenotype, would that put a stop to our drug development efforts? As you see from this post, even genes with a lethal knockout phenotype and plenty of evidence that loss-of-function variants are not well-tolerated in the human genome can still be viable drug targets. Everything depends on the details.
The code to produce the plots and analyses in this post is available on GitHub.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.