pi: PrP's hypothetical backup protein

definition
pi or π is a hypothesized protein which carries out a crucial cellular function identical to that of PrP.
history
Ever since it was first discovered that PrP knockout mice were pretty normal [Bueler 1992] despite PrP’s presence in mammals, frogs and birds, it’s been an open question why an apparently conserved protein would appear so inessential upon knockout. Bueler speculated three possible reasons, to each of which I’ll attach a one-word tagline:
- Subtlety – perhaps the knockout phenotypes are mild enough that natural selection would take generations to eliminate knockouts.
- Redundancy – perhaps another protein duplicates PrP’s function.
- Inertia – perhaps PrP’s function is no longer needed but the gene has stuck around because there’s no pressure against it.
Today we also know that PrP knockout cows [Richt 2007] appear normal, and more importantly that natural knockout goats managed to appear by chance in a dairy herd [Benestad 2012]. With regards to Bueler’s first possibility, it’s also worth noting that lab mice and dairy goats alike lead pretty sheltered lives, far away from many of the selective pressures of life in the wild. (By the way, this is true of humans in many societies too.)
The “redundancy” hypothesis gained a bit more currency several years later with the discovery that interstitial deletion mutants of PrP could cause spontaneous neurodegeneration [Shmerling 1998]. Shmerling deleted amino acids 32-121 or 32-134, thus fusing PrP’s polybasic N-terminal tail (KKRPKPGGW) to its globular C-terminal domain without the most highly conserved hydrophobic core (see alpha & beta cleavage post). This caused mice to die of neurodegeneration at age 2-3 months, without any PrPSc-like biochemical features or infectivity. What’s stranger, though, was that the phenotype was only observed when the mutant PrP transgene was expressed in PrP knockout mice. When both the interstitially deleted PrP and wild-type PrP were expressed, there was no phenotype.
Therefore Shmerling reasoned as follows. Perhaps PrP’s native function is to bind to a hypothetical receptor, dupped Lprp, in order to deliver a crucial signal without which neurons will apoptose. There exists a second protein, pi, which can also carry out this native function. In PrP knockout mice, pi binds Lprp, preventing catastrophe. In mice expressing the deletion mutants of PrP, though, the mutant PrP acts as an antagonist of sorts, binding and sequestering Lprp without activating the signal, thus preventing pi from doing its job. Co-expressing PrPC restores the signal and prevents catastrophe.
Thus the idea of pi was born. But 15 years later, no one has identified pi, and its existence no longer seems as likely as it once did. I think of it, along with PrPL and Protein X, as one of the three “lost hypotheses” of prion biology – which isn’t to say we’ve ruled it out, only that the hypothesis has been around a long time without any firm validation.
testing for pi by double knockout
The proof for a protein being pi would be that if you knock out PrP and pi, then you should get a degenerative phenotype similar to Shmerling’s mice and more severe than the knockout of either PrP or pi alone. This is what’s called a genetic interaction [Mani 2008]: when “mutations in two genes produce a phenotype that is surprising in light of each mutation’s individual effects”.
The most obvious candidate would have been Shadoo (SPRN), since it’s a paralog of PrP and is also expressed in the CNS. But double knockout of Shadoo and PrP [Daude 2012] was not much more deleterious than knockout of PrP alone – the double knockout mice had a slight, barely significant, impairment in weight gain and had only the same neuropathy symptoms late in life as have been reported for PrP single knockout mice [Bremer 2010].
Another candidate might be Doppel (PRND), PrP’s other paralog. But PRND as pi could never explain the neurodegenerative phenotype in Shmerling’s mice, since it’s not really expressed in the brain – it’s almost exclusive to the testes:
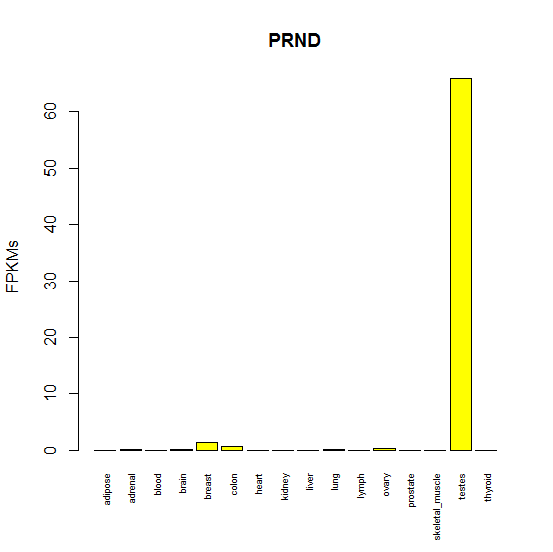
Above: barplot of Doppel (PRND) mRNA expression by human tissue, based on FPKMs from Human BodyMap 2.0 data.
And sure enough, PRND knockout proved to cause male infertility and oxidative DNA damage but it didn’t exhibit any genetic interaction with PRNP knockout [Paisley 2004].
PRND is interesting for another reason that relates to pi, though. One line of PrP knockout mice, dubbed the Nagasaki line, exhibited late-onset ataxia caused by neurodegeneration of Purkinje cells [Sakaguchi 1996]. This was attributed to loss of PrP native function and this conclusion was confirmed in a textbook rescue experiment: re-introducing a PrP transgene reversed the phenotype. But the Zurich line [Bueler 1992] and Edinburgh line [Manson 1994] of PrP knockout mice lacked this phenotype. It eventually turned out that the Nagasaki line had deleted enough material 3′ of PRNP to fuse Doppel with the remaining bit of PRNP, creating a chimeric transcript under the control of the prion promoter. This effectively led to expression of Doppel in the CNS, a phenomenon which caused neurodegeneration yet could be rescued by co-expression of PrPC [Moore 1999, Li 2000, Rossi 2001, Moore 2001].
The Doppel story is an important lesson that even the classic one-two punch of knockout-and-then-rescue can sometimes give you the wrong answer. Interestingly, it’s also consistent with the pi hypothesis. Potentially, Doppel might act like Shmerling’s Δ32-121 PrP in antagonizing Lprp and preventing pi from playing its backup role. PrPC might reverse this by binding to Doppel or by binding to Lprp with higher affinity than Doppel does.
a new deletion mutant suggests a PrP-only explanation?
It has since been discovered that deleting only amino acids 105-125 (equivalent to human PrP 106-126) is even more dramatic than Δ32-121 [Li 2007]. It causes neonatal lethality – within the first week of life – and the more PrPC you co-express, the more you delay this lethal phenotype. David Harris’ group has investigated this deletion mutant, dubbed deltaCR, in great detail. PrPΔCR is localized normally and doesn’t appear to aggregate [Christensen & Harris 2009], but rather causes non-selective currents across the plasma membrane [Solomon 2010] suggesting that this PrP mutant either blocks open a pore or is itself a pore in the membrane. This property makes cells expressing PrPΔCR highly susceptible to cationic antibiotics, a property which Harris is now exploiting in a new anti-prion drug discovery assay [Massignan 2010].
Perhaps the most fascinating aspect of PrPΔCR is that it produces these nonselective currents even in Sf9 insect cells, a cell line from army worm moths [Solomon 2010]. The relative dearth of ion channels on Sf9 cells, not to mention the dearth of potential interacting partners for PrP that would be conserved between moths and mice, makes it seem likely that PrPΔCR is not just interacting with a channel, but is itself a channel. That’s surprising because the hydrophobic domain that sometimes allows PrP to adopt a transmembrane topology [Hegde 1998 (ft)] is missing in this mutant. The PrPΔCR currents fluctuate, consistent with the idea that it takes multiple PrPΔCR molecules to form the channel, and that they spontaneously assemble and disassemble. Pentosan polysulfate prevented the currents, perhaps by binding to PrPΔCR and preventing its formation of a complex.
It’s still fairly mysterious, and Solomon speculates that the current could also result from a non-selective pore or perhaps from a “localized permeabilization of the lipid bilayer” rather than a conventional “ion channel”. In any case, though, all the evidence seems consistent with a PrP-only hypothesis to explain the toxicity of PrPΔCR and, by implication, perhaps Shmerling’s mutants as well. Pentosan polysulfate binds PrP with high affinity; so does PrP. Perhaps co-expression of PrPC prevents channel formation simply by binding to PrPΔCR. In this case, neither Lprp nor pi need be invoked.
conclusion
Of course there could still be a pi – but this construct no longer seems necessary to explain observed facts. The fact that PrPC can rescue the neurotoxicity of PrP interstitial deletion mutants might be due to direct binding of PrP to PrPΔCR. And as for Bueler’s original question, of why PrP appears conserved even though knockouts are viable, we can consider a couple of responses. First, how highly conserved is PrP really, and does conservation necessarily demand a strong knockout phenotype? I’ll address these questions in a forthcoming post. Second, the discovery of chronic demyelinating polyneuropathy (CDP) in PrP knockout mice [Bremer 2010] seems to fit Bueler’s first hypothesis, “subtlety”, quite well. Onset of CDP is in mid-life, so it wouldn’t be an absolute bar to reproduction, but it might reduce fitness enough to cause null alleles in the wild to be selected away over the course of a few generations.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.