The search for genetic modifiers of prion disease
Why genetic modifiers?
20 years ago, Yury Chernoff and Susan Liebman made a fundamental breakthrough in our understanding of yeast prions [Chernoff 1995]. In order for the [PSI+] prion to propagate, yeast must express a moderate amount of the chaperone protein Hsp104. Either knockout or overexpression of Hsp104 results in an inability to propagate prions. This apparent paradox is resolved by the breakable filament model of prion propagation. It turns out that Hsp104 is responsible for cleaving amyloid fibrils of [PSI+] to generate new ends where monomers can be added. Overexpress it, and you cleave the prions into bits too small to propagate; knock it out, and the prions can only grow linearly, adding at both ends, and not exponentially by creating new ends.
Adriano Aguzzi has opined that there must exist some chaperone or other factor in mammals required for propagating PrPSc. If there weren’t, then it should be easier than it is to generate prions de novo and propagate them in vitro. Prion propagation in vivo is like clockwork, predictable and (so far) unstoppable, while cell-free prion formation eluded researchers altogether for many years, and when it finally was achieved, has remained unpredictable and requires huge amounts of energy in the form of sonication. Do mammals have some analogue of Hsp104? What factor does so efficiently for the mammalian brain what sonication does so inefficiently in vitro?
Chernoff and Liebman discovered Hsp104 through a systematic, unbiased genetic screen - overexpressing 6000 different yeast plasmids in [PSI+] yeast to find one that would cause prion curing. Their success, and the impact that their discovery had on the study of yeast prions, are perhaps one reason why genetics has seemed an appealing approach to discover that hypothetical mammalian analogue of Hsp104. Surely discovering a protein involved in PrPSc propagation would be an enormous advance in basic science, and possibly a drug target for treating prion disease as well. And just as the possible factor(s) involved in prion propagation have remained mysterious, our understanding of pathways involved in neurotoxicity is still fairly limited too, and perhaps breakthroughs in this area could lead to drug targets for treating symptomatic-stage prion disease.
For all of these reasons, a considerable amount of effort has been expended over the past 20 years on finding genes other than PRNP that affect prion disease. In this post, I review these efforts.
Prehistory
Studies of genetic control of prion incubation time pre-date the prion concept. It was clear early on that genetically distinct mouse strains differed in their incubation times for RML scrapie [Dickinson & Mackay 1964], but these differences were eventually attributed to missense variants in the Prnp gene [Carlson 1986, Westaway 1987]. Specifically, the rare, long incubation time MoPrP-B allele differs from the reference, short incubation time MoPrP-A allele by two substitutions, L108F and V189T. This discovery that Prnp amino acid sequence differences control incubation time in mice foreshadowed similar discoveries in other organisms: for instance, that PRNP codon 129 controls prion disease risk in humans [Palmer 1991, Collinge 1991, Lee 2001] and that codon 171 controls scrapie susceptibility in sheep [Westaway 1994].
But even before the prion protein gene was established as the major basis for genetic control of prion disease risk and/or incubation time, the search for other loci had already begun. The earliest such reference that I ran across was [Kingsbury 1983]. In that study, a hypothesis about immunological differences led to a comparison of congenic mouse strains differing only in the H-2 locus (which appears to be equivalent to the HLA locus in humans). Mice differing in their H-2D haplotypes had different incubation times for RML and Fukuoka prions, and a hypothetical gene in that locus that controls incubation time was designated Pid1 for prion incubation determinant 1. However, far as I can tell, Pid1’s true identity was never determined, [Mohri & Tateishi 1989] failed to replicate an association between H-2D haplotype and incubation period, and none of the genome-wide QTL mapping studies reviewed below had peaks at H-2D. Candidate gene (or candidate locus) approaches like that of Kingsbury have continued to be popular, even as technological improvements have enabled genome-wide approaches in multiple species.
Mouse QTL studies
Traits defined in terms of continuous (height, longevity, etc) rather than dichotomous (disease vs. no disease) values are called quantitative traits. The genomic interval containing a genetic variant that modifies a quantitative trait is called a quantitative trait locus or QTL. In model organisms, QTL mapping can be performed by obtaining two distinct inbred strains of the animal which have different phenotypes. Ideally, the quantitative trait should have a tight distribution within each strain, and the difference between the means of the two strains should be large. In mice, QTL mapping can be performed by crossing two inbred mouse lines (P0) to obtain heterozygote F1s and then intercrossing these to obtain F2s which are phenotyped and genotyped.
In the early 2000s, a total of five separate studies by three independent groups sought to map QTLs contributing to differences in prion disease incubation time in different mouse strains. Here is a quick comparison of mouse QTL discovery studies:
| study | prion strain | rapid mouse background (incubation time) | slow mouse background (incubation time) | n F2s genotyped |
|---|---|---|---|---|
| Stephenson 2000 | RML | SJL/J (105 ± 4) | CAST/Ei (172 ± 6) | 153 |
| Lloyd 2001 | RML | NZW/OlaHsd (108 ± 4) | CAST/Ei (188 ± 12) | 1009 |
| Manolakou 2001 | BSE | RIII/Fa/Dk (441) | C57BL/Fa/Dk (541) | 1200 |
| Lloyd 2002 | Mouse-passaged BSE | NZW/OlaHsd (133 ± 1) | CAST/Ei (181 ± 5) | 124 |
| Moreno 2003 | C506-M3 | RIII/Fa/Dk (161) | C57BL/Fa/Dk (167) | 282 |
Note that all of the mouse strains listed above have the PrP-A allele, at least as far as the coding sequence is concerned. RIII and C57BL probably differ in non-coding regions of Prnp, as [Manolakou 2001] found linkage on Mmu2 probably mapping to Prnp. The above table does not capture all of the differences between the studies. [Stephenson 2000] and [Lloyd 2001] performed intercrosses (F1s were crossed with each other to yield F2s), whereas [Manolakou 2001] performed backcrosses (F1s were back-crossed to either of the parental strains). In addition, note that the BSE brain homogenate used in [Manolakou 2001] was a primary isolate from cattle and thus the QTLs identified could affect strain adaptation in a new species, which was not a factor in the other studies.
Each study found at least one QTL meeting the threshold for genome-wide significant linkage according to [Lander & Kruglyak 1995]. I wanted to understand how well the linkage peaks from the different studies lined up with one another, so I set out to plot them all on top of each other. I read through the papers to create a table of the microsatellite markers that were genotyped and showed some level of linkage (sadly, most of the studies did not report all markers, all the significant ones, so my data do not cover the whole genome, only the peaks), then used JAX Informatics’ marker lookup table to figure out where those markers are in relation to the modern mouse reference genome, and wrote an R script to do the matching and plot them all on top of each other. Here’s the result:
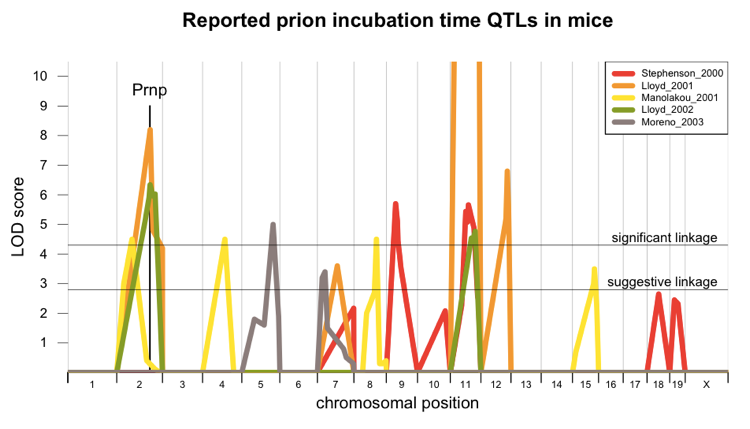
Two studies [Lloyd 2001, Lloyd 2002] found a peak that probably corresponds to Prnp itself, but the other Mmu2 peak, from [Manolakou 2001], is probably independent of Prnp. There are additional peaks on chromosomes 4, 5, 11, and 12. Three studies have agreed that there is a linkage peak on Mmu11, and in one of them [Lloyd 2001] the LOD score is off the charts (a score of about 50, orange curve cut off at the top).
The fact that the studies don’t exhibit perfect overlap isn’t necessarily an indication of anything wrong. While the underlying genes that are capable of modifying prion disease are present in all of these mouse strains, whether or not a given QTL study can actually detect such a gene depends on whether there are functional polymorphisms between the two mouse strains that affect gene expression or function. Since these studies used different mouse strains, they didn’t all have the same genetic differences between strains to work from.
QTLs in and of themselves aren’t all that interesting - the ultimate goal is to find the specific gene and variant that give rise to the observed linkage. Consider that for instance all of the studies used i.c. inoculation as the method of prion infection. One can therefore imagine many plausible mechanisms by which genes might influence incubation time - some could affect prion replication or degradation, which are mechanisms we would be very interested in from both a basic science perspective and a human health perspective. Others might conceivably affect initial clearance of foreign inoculum, which would not be relevant to sporadic or genetic prion diseases in humans and probably wouldn’t tell us anything very fundamental about prions either. Only when a quantitative trait variant has been identified can one proceed with the functional studies that would clarify how the gene affects prion disease mechanistically.
For this reasons, the QTLs plotted above therefore led to several follow-up studies aimed at identifying the causal gene. For instance, George Carlson and Lee Hood pursued their study of differential gene expression over the course of prion infection [Hwang 2009] in part to identify the most promising candidate genes under the linkage peaks. In light of the linkage at Mmu2, and particularly after mahogunin was proposed as a possible mediator of neurodegeneration prion disease [Chakrabarti & Hegde 2009], George Carlson tested whether knockout or overexpression of Atrn or Mgrn1 would affect incubation time - neither did [Silvius 2013, Gunn & Carlson 2013]. Meanwhile, MRC Prion Unit pursued further fine mapping on Mmu11 and 15 and examined the candidate genes Mcp1 and Cpne8 respectively. Today, cheap genotyping and sequencing technology would make it possible to do these sorts of fine mapping studies in a relatively more comprehensive and unbiased manner, but these studies were done several years ago. Because Mcp1 knockout had already been reported to extend incubation time for ME7 prions [Felton 2005], it was considered as a candidate to explain the Mmu11 linkage peak, but when Felton’s results failed to validate in an RML-infected model [O’Shea 2008] the peak was not considered further, as far as I can tell. The linkage interval on Mmu15 contained 39 genes, but (presumably due to sequencing cost) only 29 of them were genotyped to identify candidate polymorphisms. One gene with a large number of polymorphisms, Cpne8, was considered as a candidate gene, and shown to be differentially expressed in prion infection [Lloyd 2010], but knockout/overexpression studies were never conducted.
Genetic studies in livestock
Several studies have looked for genetic linkage in sheep and cattle. I found these studies interesting because the methods were sort of a hybrid of other things I’ve learned about. Mouse genetics is interventional - set up the cross, breed the mice, etc. - while human genetics is observational - you have to do what you can with the humans you’ve got. Livestock are somewhere in between - they’re sufficiently more expensive than mice that you’re somewhat more limited to observational study. Yet they have an inbred population structure more like that of laboratory mice.
Two studies have looked for quantitative trait loci in sheep, though with a slightly different study design than in the mouse studies above. One study followed two half-sibships (offspring all born of the same father or same “sire” as they say in sheep parlance) of ARQ/VRQ sheep on a scrapie-infected farm and tracked their scrapie incubation times [Moreno 2008]. It found linkage peaks that were “chromosome-wide significant” in one half-sibship at the .05 level on OAR6 and OAR18, but no peaks that were genome-wide significant. A subsequent followup study [Moreno 2010] genotyped and phenotyped a further >500 sheep of various pedigree structures on the same farm. This study found additional evidence for linkage on OAR18, though it is not clear to me whether this was genome-wide significant. There was no support for linkage on OAR6.
One study performed transmission disequilibrium tests on cattle with BSE [Hernandez-Sanchez 2002]. Another series of studies performed genome-wide association studies on Holstein cattle, in a case (BSE) vs. control (no BSE) study design [Murdoch 2010, Murdoch 2011]. The cattle genome is about the same size as humans - 3 Gb - though I don’t know in terms of centimorgans, which would better represent what threshold should be used for genome-wide significance. In any case, these studies have identified some suggestive peaks but the lowest p value reported so far is 5e-5, so not genome-wide significant.
Genome-wide association studies in humans
The first GWAS for prion disease in humans used 929 prion disease patients and 4,254 controls, focusing primarily on vCJD [Mead 2009]. This study confirmed the known association between codon 129 and vCJD risk, and also identified other SNPs in and around PRNP that were still associated with risk after conditioning on codon 129. It also noted a SNP in RARB (retinoic acid receptor beta) with a p value of 1.9e-7, somewhat short of the canonical genome-wide significance threshold of 5e-8. This possible association with RARB was further studied by performing a candidate QTL analysis (a cross of 1000 heterogeneous stock mice and then targeted sequencing of candidate genes) in prion-infected mice, which showed a nominal association between Rarb genotype and incubation time (p = .0005) [Grizenkova 2010]. To my knowledge, knockout/overexpression studies of Rarb have not been reported.
The second GWAS came a few years later and included about 2000 cases and 6000 controls [Mead 2012]. The cases included acquired, genetic and sporadic prion disease, so there were a number of different subgroups to be dealt with. Interestingly, an allelic model - which cannot capture the enormous heterozygote advantage at codon 129 - still picked up SNPs in PRNP, probably related to a very small difference in risk between 129MM and 129VV genotypes. In contrast to the 2009 GWAS, this study did not find residual association at PRNP after conditioning on codon 129 genotype. It also did not find any other genome-wide significant loci.
That picture may now be changing. Simon Mead announced at Prion2014 Day 4 that his number of sporadic CJD samples has more than doubled, and he now sees several apparently genome-wide significant (p < 5e-8) hits. He is currently seeking any investigators with appropriately consented DNA samples from prion disease patients to contribute to a replication study before publishing these findings.
Knockout and overexpression in mice
Arguably, the ultimate test of whether a mouse gene affects prion incubation time is whether knockout or overexpressing mice have different incubation times than their wild-type littermates do. Additionally, we might hope that if the association is real, then knockout and overexpression should have opposite effects. But that wasn’t true in the case of Hsp104 in yeast, so we should also stay open-minded if there is strong evidence for knockout and overexpression both giving real effects and in the same direction.
To date, tens of genes have been knocked out and/or overexpressed in an effort to determine whether they affect the course of prion disease. Such studies have identified genes that dramatically affect the ability of peripherally acquired prion disease to invade the CNS - for instance, complement receptors CD21/35 [Michel 2012]. However, studies of i.c. inoculated mice have given largely negative results, and the few positive results that have been reported are of very limited effect size. In the table below, I have tried to collect all of the candidate gene knockout and/or overexpression studies from i.c. inoculated mice. Blank cells indicate the experiment has not been reported.
| mouse gene | knockout affects prion disease? | overexpression affects prion disease? | citations |
|---|---|---|---|
| Aplp2 | no | [Tamguney 2008] | |
| Apoe | no | [Tamguney 2008] | |
| App | yes | no | [Tamguney 2008] |
| Atrn | no | [Gunn & Carlson 2013] | |
| Cav1 | no | [Tamguney 2008] | |
| Ccr2 | no | no | [Tamguney 2008] |
| Ccr5 | no | no | [Tamguney 2008] |
| Cd9 | no | [Tamguney 2008] | |
| Dpl | no | [Behrens 2001, Tamguney 2008] | |
| Fyn | no | no | [Tamguney 2008] |
| Hectd2 | no | no | [Begum 2013] |
| Hsp70 | no | [Tamguney 2008] | |
| Il10 | no | [Tamguney 2008] | |
| Il1r1 | yes | [Tamguney 2008] | |
| Mapt | no | [Lawson 2011] | |
| Mcp1 | supposedly | [Felton 2005] | |
| Mgat3 | no | [Tamguney 2008] | |
| Mgrn1 | no | no | [Silvius 2013] |
| Msra | no | [Tamguney 2008] | |
| Msrb | no | [Tamguney 2008] | |
| Nox2 | yes | [Sorce 2014] | |
| Ptpra | no | [Tamguney 2008] | |
| Sod1 | yes | [Tamguney 2008] | |
| Sprn | no | [Daude 2012] | |
| Tgfb1 | no | no | [Tamguney 2008] |
| Tnf | no | [Tamguney 2008] |
Positive results were reported for App, Sod1 and Il1r1, but opposite directions of effect between knockout and overexpression were not shown, and these genes do not seem to have been pursued further. The most convincing result is the recently reported effect of Nox2 (NADPH oxidase) knockout [Sorce 2014] - although the effect size was very small, it seems to indicate some role for oxidative stress in prion neurotoxicity.
Reading through these studies, it is haunting to see how much effort often went into chasing up candidate genes for which no convincing effect on prion disease was ultimately demonstrated.
For example, one travels a very long road from the originally reported phenocopy of prion disease in mahoganoid mice [He 2003] to the reported depletion of mahogany by cytosolic PrP [Chakrabarti & Hegde 2009] to finally find that mahogany (Mgrn1) knockout and overexpression do not, after all, affect prion disease [Silvius 2013].
Another well-documented story is that of Hectd2. Although one study [Stephenson 2000] found an almost-suggestive QTL on Mmu19, the chromosome where Hectd2 is located (see plot above), this genomic region was actually subjected to fine mapping for a different reason, which was “an interest in candidate genes on human chromosome 10 (unpublished data)” [Lloyd 2009]. Fine mapping and sequencing of candidate exons and UTRs in a heterogeneous mouse stock of mice led to an interest in Hectd2. Mouse strains with short incubation times (AKR and BALB/c) had 2.4x higher whole brain Hectd2 mRNA levels by qPCR than strains with long incubation times. A minor allele of the human ortholog was nominally (p = .0049) enriched (odds ratio of 2.11) in vCJD patients compared to population controls (the possibility of population stratification is not discussed in any detail), and minor allele heterozygotes had 2.3x higher HECTD2 mRNA levels in blood than homozygous reference humans. If Hectd2 were the quantitative trait gene, then these findings would lead one to hypothesize that Hectd2 knockout would extend incubation time, while Hectd2 overexpression would result in a more rapid disease course. Subsequent knockout/overexpression studies have failed to validate any association between Hectd2 and prion incubation time [Begum 2013]. A total of 22 comparisons are made between knockout vs. wild-type and overexpresser vs. wild-type mice (Table 6.3.1 and Table 6.5.1), with virtually identical times to endpoint in almost all of them. The only one comparison that remains significant after multiple testing correction is that of time to first symptoms in knockout vs. wild-type females inoculated with RML prions, but this is not highly convincing given the “±0” standard deviation, lack of replication in other prion strains, sexes or endpoints, and the fact that the supposed direction of effect (knockouts had shorter incubation time) contradicts the original study [Lloyd 2009].
Given the widespread tendency not to report negative results, I suspect that the above stories represent just a fraction of all the efforts that have gone into determining whether various genetic manipulations can affect prion incubation time. Depending on your perspective, the preponderance of negative results in this area might be either a reminder that most things don’t work in science, period, or a reminder that nominal associations and seemingly compelling biology do not provide sufficient basis for choosing candidate genes, and that instead what we really need are better genome-wide approaches.
Outlook
The papers cited in this post represent a huge number of animals, a huge number of dollars and a huge number of hours of work. For all this effort, we so far have very little idea what, if any, genes besides PRNP affect prion propagation and neurotoxicity. The association with Nox2 [Sorce 2014] appears to be real, and the associations for App, Sod1 and Il1r1 might or might not be real. And the possibility, however speculative, of genome-wide significant hits from the newest generation of GWAS is exciting as well. But the overall impression I am left with after reviewing all of this literature is that we need bigger, better, genome-wide approaches.
Luckily, that seems to be the direction in which biology is moving. CRISPR loss-of-function screens appear to present an opportunity to do genome-wide mutagenesis screens in cultured mammalian cells, of the type that were once possible only in yeast or (with great effort) maybe worms and flies. The challenge is finding a cell culture model that is relevant to the in vivo situation.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.