Genetics in medicine 08: Inflammatory bowel disease
These are my notes from week 8 of Harvard’s Genetics 228: Genetics in Medicine from Bench to Bedside course, held on March 27, 2015. Lectures by Mark Daly, chief of the Analytical and Translational Genetics Unit at MGH and Ramnik Xavier, chief of Gastroenterology at MGH, and a journal club presentation by me, Rebecca S. Fine and Vinita Joshi.
Background
For journal club today, my group is presenting the paper Genetic and epigenetic fine mapping of causal autoimmune disease variants [Farh & Marson 2015]. Here’s a quick backgrounder.
Successes and limitations of genome-wide association studies
The concept of a genome-wide association study (GWAS) is introduced here. The first GWAS is sometimes credited to be a study of 116,000 SNPs in 96 cases and 50 controls for age-related macular degeneration in 2005 [Klein 2005]. But it was not until a couple of years later that GWAS really hit their stride, reaching the scale needed for locus discovery in common diseases, when the Wellcome Trust Case Control Consortium genotyped 17,000 people on 500,000 SNPs [WTCCC 2007; see also historical review in Visscher 2012]. Since then, over 1,700 GWAS have been published, reporting associations for over 11,000 SNPs [Welter 2014].
A few general trends have emerged. Only about 11% of GWAS loci are in protein-coding regions [Gusev 2014], which is a dramatic enrichment (as only about 1% of the genome is protein-coding), but still means that the vast majority of causal variants must be non-coding. 57% of GWAS hits lie in open chromatin (DNAse I hypersensitivity regions) [Gusev 2014], and expression quantatitive trait loci (eQTLs; variants that affect gene expression) are enriched among GWAS hits [Nicolae 2010]. Thus, much of the functional impact of causal variants might lie in their effects on gene expression.
Still, there are only rare cases where people have been able to confidently identify a single variant as driving the GWAS association at a particular locus. The reason that GWAS work at all is also their greatest weakness. Recombination hotspots effectively divide the human genome into haplotype blocks of 2-200kb [International HapMap Consortium 2003 and also see here], within which genetic variants almost always segregate together. Suppose a particular 20kb haplotype block contains 20 SNPs, one of which is causally associated with your phenotype of interest. Then genotyping any SNP in that block (or even nearby) will reveal that association. That’s why GWAS work - you don’t need to genotype the causal SNP to discover an association. But this also makes it incredibly difficult to tell which of those 20 SNPs is causal. And until you know the causal variant, it can be hard to reach the sorts of biological insights that will lead to therapeutic avenues or fundamental basic science insights.
Therefore people have devised a lot of strategies for trying to at least narrow down the set of causal variants at a phenotype-associated locus. One approach relevant to this paper is fine-mapping. This is when you re-genotype the same people (or more people, if you have them!) with a chip that has a much denser array of probes at the causal locus. For instance, the ExomeChip, Immunochip or PsychChip. Why didn’t we just put all of those SNPs on the GWAS array in the first place, you ask? Capacity on chips isn’t free or infinite, and GWAS discovery chips tend to prioritize uniform coverage of the genome and very high quality, readily genotyped SNPs. These fine-mapping chips instead try to exhaustively capture variants found in sequencing studies such as 1000 Genomes or ESP, even if it means some of the probes are of lower quality and do not uniformly cover the genome. Even with fine-mapping, you’ll still end up with a lot of variants at one locus all associated with your phenotype, so this is by no means the end of the story. If you can’t afford to re-genotype your study participants or don’t have any DNA left, you can try to guess what other SNPs they have in between SNPs you’ve genotyped, based on the haplotype structure of 1000 Genomes participants - this is called imputation. This is a substitute for fine-mapping and has all the same limitations, plus its own limitations - you won’t guess genotypes correctly 100% of the time.
So, as a very simplified example to show how these things work, consider this imaginary locus (code to generate this plot is here):
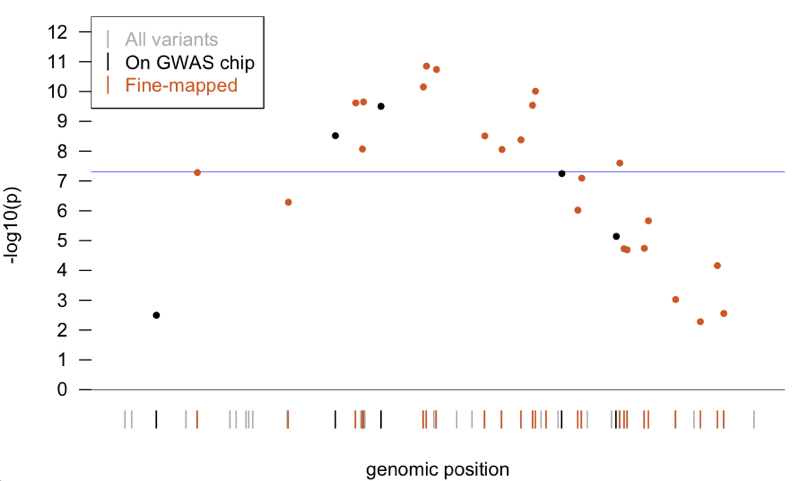
You’re interested in figuring out, of all the genetic variants in the population (gray bars), which are associated with your phenotype. As a first pass, you genotype a bunch of cases and controls for only a few, relatively uniformly distributed, well-genotyped SNPs (black bars). You find a few SNPs nominally associated, and two SNPs that are significantly associated with your phenotype - they’re above the genome-wide significance threshold of p = 5×10-8. So then you go back with a fine-mapping chip, and you genotype much more (but still not all) of the variation here (orange bars), and you find a bunch more SNPs that are associated, including several that are above the genome-wide threshold and a few that are more significant than your original GWAS hits. But you still don’t know which, if any, of the SNPs you’ve genotyped is causal, so how can you make any biological inferences? That question is where this paper picks up.
The genetics of autoimmunity
Of all the diseases and phenotypes that have been subjected to GWAS, autoimmune diseases must rank among either the most challenging or the most successful, depending on your viewpoint. I haven’t found an updated figure for 2015, but as of three years ago, there were already 193 independent genomic loci associated with inflammatory bowel disease (IBD) [Jostins 2012]. This degree of polygenicity is so vexing that it has motivated whole new statistical approaches to determine whether the associations are largely real or result from confounding effects of population stratification [Bulik-Sullivan 2015]. Specific functional variants have been identified in a handful of loci - see for instance CARD9 [Rivas 2011] - but for many or most of these loci, we don’t know how the genetic variation impacts gene function, and we don’t know if increased or decreased gene expression would associate with disease risk.
Mark Daly - Crohn’s disease: gene discovery and beyond
Dr. Daly phrases the goal of human genetics as:
To provide durable insights into human disease biology that ultimately impact clinical care.
In most areas of epidemiology, correlation does not imply causation. The power of human genetics lies in the fact that correlation does imply causation - you are born with your genome and get the disease later; the disease does not change your genome.
A secondary focus of human genetics might be to deliver individualized medical insights - diagnosis in severe genetic disorders, identification of individuals predicted to respond besst to a therapy, prediction of adverse drug responses. The potential for these sorts of personalized insight has been dramatically oversold for years, and for the most part has not lived up to the hype, though we may now be nearing the time when some of these promises do become reality.
Linkage mapping was developed by Alfred Sturtevant and Thomas Hunt Morgan in 1913, as a purely mathematical concept decades before we knew the structure of DNA, or the genetic code, or had any tools for experimental manipulation of DNA. It was also recognized early on that certain human diseases - Huntington disease, cystic fibrosis, muscular dystrophies - were inherited in a Mendelian fashion. In the early 1980s it became possible to track the co-segregation of DNA markers with these diseases, and by the late 1980s this began to enable the identification of causal genetic variants in many of these diseases. This was possible because there is a deterministic relationship whereby genotype causes disease state.
By 2002 there were ~1800 Mendelian diseases for which the causal variant had been found, but fewer than 10 known complex trait genes [Glazier 2002]. It had been known for a century that there are many traits that are highly heritable - height, for instance, may be ~80% heritable. Yet for many traits, finding the genes that determine them was nearly impossible. In 1918, the statistician R.A. Fisher put forth the possible explanation that such traits might be controlled by a large number of genes that did segregate in a Mendelian fashion but each had only a small effect size [Fisher 1918].
In the past decade, a new approach has become feasible. We can systematically compare DNA variation between two groups of samples. For instance, in microbiology we can compare antibiotic resistant and sensitive bacteria to identify the variants that confer resistance. In cancer we can compare tumor and normal DNA to identify mutations that drive cancer. And in common diseases and traits, we can compare individuals with or without the disease to find the variants that control the trait.
Soon after the Human Genome Project was completed, it was recognized that human heterozygosity is relatively limited (~1 base pair in 1000), and most of it could be attributed to common genetic variants [Gabriel 2002]. When you discover a SNP that is heterozygous in one individual (differs between two chromosomes) and then go out and genotype that SNP in a thousand other people, about 90% of the time you discover that the variant has a frequency of >1% in the population.
It turns out that humans are not very different from one another. In fact, we are among the least diverse organisms on the planet. Almost all species ever studied have a greater amount of DNA variation than we do. We began to catalog the structure and frequency of genetic variation in humans. There are only about 10 million common genetic variants in humans, and most of them arose from a single mutational event in our history. The data also resoundingly confirmed the anthropologists’ “out of Africa” hypothesis that humankind had originated in Africa. The mapping of Mendelian disease genes actually relies on common variants. The evidence that the cystic fibrosis gene was on chromosome 7 came from the observation that over half of CF patient chromosomes had an identical common variant haplotype of about 500kb. Usefully, recombination events tend to occur at specific hotspots, thus creating haplotype “blocks” of tens or sometimes hundreds of kb, within which there is almost never recombination [Daly 2001] - thus, common variants within a block segregate together.
Once all of these discoveries were made, three fundamental changes came about that enabled common disease genetics:
- Large consortia - first the SNP Consortium, then HapMap and then 1000 Genomes
- Advances in technology - first genotyping arrays, then sequencing
- Collaborations among disease researchers - more on this later
Inflammatory bowel disease appeared to have many advantages for common trait genetics. It is highly heritable: it has 15-35x risk in siblings of affected individuals, much higher monozygotic than dizygotic twin concordance, and 5-10% of affected people have a 1st degree relative who is also affected.
Early linkage studies enabled the 2001 discovery of NOD2 at 5q31 which explained 2-3% of the heritability of inflammatory bowel disease. Then all of the years from 2002 to 2008 were spent on candidate gene studies which yielded no new validated hits. Finally, by 2008, genome-wide association studies (GWAS) became possible. However, it soon became clear that GWAS were different from anything that had come before them in molecular biology. In all of molecular biology up to that point, you got a gel or some sort of readout that you could look at with the naked eye and decide whether you believed the data. With GWAS you could now get a volume of data that no one human would ever look at with the naked eye. Thus, we had to develop:
- Technical QC - removing failed SNPs and samples
- Genetic QC - Mendelian segregation, Hardy-Weinberg equilibrium, estimating relatedness and gender, controlling for population structure
- Analytical QC - checking for inflation of test statistics, biases toward missing data, etc
Those QC efforts were 95% of the labor. The actual effort of testing for association is remarkably simple. You basically just make a contingency table of allele A vs. allele C in cases vs. controls and do a Chi-square test.
In the first Crohn’s disease Gwas, after about 500 cases and 500 controls, the highest-ranked SNP had a p value on the order of 1e-7. At first, this looked good because in linkage studies in families, a LOD score of ~3 was considered significant, because there are only so many arms of chromosomes, only so many recombination events in one family. But quickly they realized that because large populations have had many recombination events and have a 3.2Gb genome broken into, say, 3.2kb blocks, we are really doing more on the order of one million independent statistical tests in a genome. Thus, p = .05 × 1/(1 million) = 5e-8 became the standard threshold for genome-wide significance. Moreover, people decided that they almost never believe a single association study - only when an independent technology and independent collection of cases and controls have been used to replicate the result does it become real.
That original hit in Crohn’s disease was not real, but soon they had 1000 cases and 1000 controls, and found two genuine genome-wide significant peaks, one at NOD2 which was already known since 2002, and one at IL23R which turned out to be a coding variation.
Two plots became standard in conveying the results of genome-wide association studies: the Manhattan plot and the Q-Q plot. The Manhattan plot is mostly just eye candy and (usually) doesn’t tell you much about the strength of statistical evidence for association with disease. The Q-Q plot is important because if the associations are real, it will show you a distribution of p values perfectly consistent with expectation (no inflation), except at the very high end where your real hits are.
To get the numbers needed to find a large number of hits, you really needed more samples. Thus, the third fundamental change (above) was that disease communities - which used to compete to find the one causal gene in Mendelian disease - realized that there were many genes to be found and they could afford to collaborate to find them all. Therefore, people started to combine data from multiple GWAS studies in different case and control cohorts genotyped on different technologies. The different technologies would genotype a different set of SNPs, which meant that to compare them it was necessary to impute the missing SNPs on each chip using the HapMap reference panel. Once this was done, the first meta-analysis in 2008 revealed that simply by combining data from three already-published studies, you could find many new loci that were not significant in any one study.
The findings of GWAS have proven impressively durable. All 32 of the GWAS hits from the first IBD meta-GWAS [Barrett 2008] have been confirmed and are actually more significant in the 2012 GWAS data [Jostins 2012].
Does it matter that all of the GWAS hits have very small influences on the disease traits? No, because the (small) effects of common variation do not put a limit on the (large) potential effects of a drug. For instance, common variation in HMGCR has only a very small (~5%) influence on LDL levels, yet inhibition of HGMCR with statins has a large (~30%) effect on LDL. Human genetics is constrained by natural selection - common variants (say, HMGCR) have to have small effects, while variants of large effect (say, LDLR mutations) are very rare. But drugs are under no such constraint - you can develop a drug with a large effect, and give it to a large number of people.
The major challenge coming from GWAS is how to identify the exact variants that drive GWAS associations and, mechanistically, how they affect disease risk. There are a few success stories - for instance, null alleles of FUT2 confer norovirus resistance but also increase IBD risk - but more often we do not know why a given locus affects disease risk, or even which gene is involved.
Many of the most valuable insights from GWAS have been from the overlap of associations between one disease and another - for instance, the overlap of IBD loci with leprosy GWAS and with Mendelian primary immunodeficiency genes has told us a lot about the biological pathways shared between these conditions. Similarly, it has been useful to discover the concordance (or lack thereof) between distinct but clinically similar diseases. Ulcerative colitis and Crohn’s disease share many but not all of their loci, and a polygenic “risk score” based on genotypes at these loci adds a considerable amount of diagnostic accuracy (when combined with the conventional smoking and serological status meausres) at discriminating the two diseases. The patients that had been mis-classfied were almost categorically those who had responded the least well to treatment, so re-assessing their diagnosis using genomics is important.
Promisingly, exome and genome sequencing now make it possible both to find rare, more highly penetrant, genetic variants, and to identify causal common variants at GWAS loci. An example success story was the discovery of homozygous essential splice site mutations in DGAT1 in two children with a congenital severe protein-shedding enteropathy. These sorts of insights are valuable both personally - for genetic testing and in some cases for guided therapies - and also for fundamental biology. DGAT1 is an example where the knockout phenotype is very divergent in mice and in humans. Dgat1 knockout mice have very low lipids or something, so millions of dollars had been invested in developing drugs to inhibit DGAT1, but these caused a very high rate of adverse side effects. The discovery of this severe DGAT1 loss-of-function disease in humans explained why that was the case. If we had been guided by human genetics to begin with, we wouldn’t have made that mistake.
Indeed, being guided by human genetics can lead to promising new drug targets. SCN9A (Nav1.9) knockout causes congenital indifference to pain [Cox 2006], and gain-of-function mutations cause extreme pain disorder. There are now small molecule inhibitors of Nav1.9 [Yang 2013]. Another great example is PCSK9. In IBD, the most promising genetic result is that a rare splice variant that abolishes part of the protein [Rivas 2011] confers dramatically reduced risk of IBD, motivating an effort to find a drug that would mimic the effect of that genetic variant.
Q&A
Q. Doesn’t it seem like hits that are just barely above the 5e-8 threshold turn out to be false more often than 1 in 20 times?
A. Actually, hits that are just below 5e-8 later turn out to be real much more often than you’d think. This is for a Bayesian reason. Now that we know that many of the diseases we study in GWAS are so polygenic, there is a strong prior that a real association would be the one that “happens” to be at the high end of the null distribution, at say 1e-7 or something. However, we chose the 5e-8 threshold to be conservative because back in the linkage era we hadn’t been conservative enough. Linkage studies are only powered to find variants of large effect, yet people had applied them to diseases where the prior for common variants of large effect was extremely small. Therefore thousands of people would do those linkage studies, and 1 in 20 would find (and publish) a positive result, all of which were false.
Ramnik Xavier - Inflammatory bowel disease
IBD is divided into ulcerative colitis (UC), Crohn’s disease (CD), and “intermediate colitis” (intermediate phenotype, 10-20% of patients). Each can be divided into several subtypes and it is possible that we will eventually consider them distinct diseases. Each of these diseases is rapidly progressive, with inflammation spreading across the colon in a matter of 2 weeks. UC typically presents first with bleeding, while CD presents first with abdominal pain resulting from stricture preventing flow through the bowels. In some cases, ulceration can reach all the way through to the skin, resulting in visible fistulas.
Ulcerative colitis usually has the same histological profile, can follow approximately four very different disease courses [Solberg 2009] - some patients have one bout, then a remission of >10 years; others have recurrence every 5 years; others have constant disease; and so on.
Treatment outcomes are quite poor. IBD does not respond to steroids (unlike other inflammation). Children experiencing their first bout do have longer remission if treated with 6-mercaptopurine. There are several known environmental factors. Smoking makes ulcerative colitis better but Crohn’s disease worse.
IBD is an interesting disease because of the interaction between genetics, immunity and microbial flora. Before 2002, people had theories that we’d find a specific gut pathogen that triggers IBD - that never happened, but we have now gained a more nuanced understanding of the ways that the gut microbiome does influence disease risk.
The RISK cohort was a combination of 28 pediatric IBD centers to sample gut microbiomes of 1300 children with Crohn’s before treatment, during their first attack. Microbes are identified by sequencing their 16S RNA. The “good” bugs (especially Clostridiales) were decreased in cases vs. controls [Gevers 2014]. Sadly, treatment with antibiotics - which many parents of kids with IBD demand and physicians give in - further depletes the “good” bugs and results in far worse outcomes. There are now efforts to develop a therapeutic microbe supplement full of the good bugs.
One key pathway that has emerged from IBD GWAS is the TH17 pathway, specific to TH17 cells. GWAS loci at IL23R, RORC, CCR6 , STAT3, and Aiolos (what is that??) all point to this pathway. An early hypothesis to explain the IL23R cells was that the protective variant reduced the number of circulating TH17 cells. Instead, it turns out the number of cells is the same but they secrete less TH17 protein.
Another revelation from IBD genetics has been that IBD genes fall into three very broad categories: those that control homeostasis, those that control inflammation, and those that control healing (affecting recovery after an attack of inflammation).
An ongoing effort is to do extreme phenotype exome sequencing - for instance, looking for protective rare variants in people who have a high polygenic risk score based on common variants but who do not have the disease.
Prior to 2007, there were no references on Crohn’s disease and autophagy. Beginning in 2007, two GWAS identified ATG16L1 T300A [Hampe 2007] and a 20-kb promoter deletion upstream of IRGM as being associated with IBD. Both of these are autophagy genes. Atg16 (encoded by ATG16L1) is a core component of the autophagosome machinery, but the T300A missense variant is in the C-terminal domain which is evolutionarily more recent. The N-terminus of ATG16L1 has a homologue in yeast, but the C-terminal domain does not and thus probably is not involved in the protein’s core conserved activities, but rather in some higher-order function. It turns out that the risk allele of ATG16L1 is much less effective at clearing bacteria in culture [Kuballa 2008], and T300A knock-in mice have fewer Paneth cells and enlarged goblet cells [Lassen 2014].
It is increasingly clear that abnormal Paneth cells are granuloma-poor and have a reduced ability to contain pathogens, and these abnormal cells are much more often found in patients with NOD2 or ATG16L1 risk variants, possibly defining a new subtype of Crohn’s disease [VanDussen 2014].

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.