Results in from PRN100 antibody first-in-human dosing
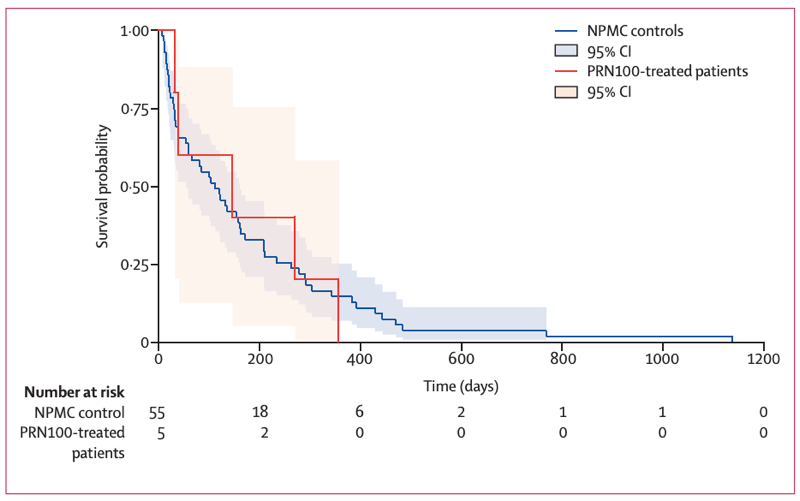
Figure 2 from [Mead 2022], showing the survival of patients treated with a PrP monoclonal antibody (red) versus historical controls (blue).
MRC Prion Unit in London has just published the results of its first-in-human dosing of the PRN100 antibody against PrP [Mead 2022]. For those just tuning in now, this antibody has been in development for more than 20 years [White 2003], and after many years of preparation, human dosing finally began about 3 years ago.
background
It was discovered in the early 2000s that monoclonal antibodies to PrP could clear prion infectivity in cell culture [Enari 2001, Peretz 2001]. An important in vivo study in mice then delivered a complicated result. When the monoclonal antibody ICSM18 was given intravenously to mice infected peripherally with prions, before the infection had migrated to the brain, disease could be delayed out to at least 500 days, perhaps indefinitely [White 2003]. But when the same treatment regimen was given to mice infected directly into the brain, or in whom the peripheral infection had already reached the brain, no benefit was observed. This treatment of mice with a brain prion infection might have failed because it was too late, because not enough drug got into the brain, or for some other reason. This left open the question: could antibody therapy targeting PrP successfully treat a prion infection in the brain? In the two decades since then, no study in animal models has delivered a convincing answer to this question [Song 2008].
Aspiring to advance ICSM18 as a drug candidate, MRC Prion Unit humanized the antibody — the human version is called PRN100 — and manufactured a clinical-grade batch of it. Of that initial batch of drug produced, some was subjected to toxicology studies in mice and monkeys to determine safety and pharmacokinetic parameters. Although the antibody’s developers hoped to launch a formal clinical trial, years went by and this never came to fruition. Instead, as I explained in detail in my last post on this subject, PRN100 eventually came to be administered to humans under what the U.K. government calls a Specials Licence or a Specials Exemption. Whereas a clinical trial is designed as a research study — a science experiment, really — here the treatment was administered as an emergency compassionate use therapy. That meant that the primary goal was treatment, not research. Patients were not randomized, and indeed, patients could not be subjected to blood draws or lumbar punctures solely to obtain fluids for research. So the studies of the drug’s pharmacology were limited to whatever leftover samples were collected in the course of clinical care. The study was also limited (presumably for financial or manufacturing reasons) to whatever remained of that initial batch of drug, so there was only enough to dose six patients, of whom two had to stop treatment when the drug supply ran out. While the primary goal was treatment of patients, we are all eager to learn anything we can about what a PrP monoclonal antibody treatment might do in a prion disease patient, and so the MRC Prion Unit did all the research that it could based on the available patient data and samples, and has now written that up as a publication.
summary of the study
The 6 patients were started on PRN100 a range of 1-11 months after their onset of disease. They were already impaired, but not at end stage: scores on the MRC Scale, which measures ability to carry out basic activities of daily living [Thompson 2013], with 20 being unimpaired at the time of therapy initiation, averaged 14.7 (range 11-18). They received PRN100 as a 4-hour IV infusion, starting at a 1 mg/kg dose and escalating eventually to a 120 mg/kg dose repeated every 2 weeks. Because patients progressed at different rates and died or withdrew, and the drug supply eventually ran out, not every patient ended up being on a sustained high dose for any length of time.
No serious adverse events were observed upon drug treatment. All of the patients eventually died of prion disease, and overall, their survival time and their progression along the MRC Scale looked no different from historical controls (see image at top). One patient did have a longer survival time and slower progression relative to historical controls from their same subtype (HGH iatrogenic CJD), although that individual had already exhibited slower progression than those controls before PRN100 treatment began.
Peak serum drug concentrations reached 13-35 μM and peak CSF drug concentrations reached 16-132 nM, for a CSF:serum ratio of at least 0.11%. In the one individual who died 3 days after their final dose of drug and had autopsy done after an additional 9 days, drug concentration in the brain parenchyma was measured at 9.9 - 27.4 μg/g across several brain regions. (If one assumes that a gram of brain tissue is about 1 mL of volume and that an IgG antibody is about 150 kDa in molecular weight, that corresponds to a concentration of 66-183 nM).
The authors concluded that the positive safety profile of the drug combined with evidence of potentially meaningful drug concentration in CSF and brain would justify a formal Phase II clinical trial of PRN100 in symptomatic prion disease patients, with a potential to eventually expand into healthy at-risk individuals.
analysis and discussion
As expected since this was not a clinical trial, the data from the limited human dosing of PRN100 are unable to answer the question of whether PRN100 can safely and effectively treat prion disease. On one hand, the survival times and rates of disease progression in the six treated individuals were not obviously different from the best-matched historical controls. On the other hand, 6 patients is not nearly enough to provide statistical power to determine whether these outcomes were modified, and perhaps just 3 or 4 of these individuals actually underwent a meaningful duration of treatment at the highest dose. Instead, as I foreshadowed in 2019, the overall survival and progression data from this effort so far only rule out the most extreme outcomes: the antibody did not cure prion disease, nor was it severely toxic. Beyond disease outcomes, though, what can this experience teach us?
An open question in the development of monoclonal antibody therapies for brain diseases generally has been if, and how, one can get enough drug into the brain. The dosing regimen in this study was aggressive, escalating as high as 120 mg/kg biweekly. A typical adult human might weigh 50-80+ kg, so that’s 6-10 g of drug per 2 weeks, hence the need for a 4-hour IV infusion. For comparison, the antibody adalimumab, currently the best-selling drug in the United States, is labeled by FDA for treatment of arthritis, psoriasis, inflammatory bowel disease and other conditions at a maintenance dose of 40 mg total (not per kg) every 2 weeks. Aducanumab, the controversial Alzheimer’s drug, is labeled at a dose of 10 mg/kg (so, say, 500 - 800 mg total) every 4 weeks. The highest dose I’ve heard of for an antibody in trials for a brain disease is solanezumab, at 1600 mg every 4 weeks. (A cursory Google search revealed no precedents for clinical testing of higher doses of antibodies.) Thus, the PRN100 dose here is perhaps 250 times the adalimumab dose, 25 times the aducanumab dose, and 10 times the highest dose I’ve ever heard of in clinical testing. It is remarkable, then, this dose appeared to be tolerated, with no serious adverse events and no indication of symptoms similar to ARIA, a side effect that has long plagued Alzheimer’s Aβ antibody trials [Sperling 2011]. (The supplement describes briefly some of the animal toxicology studies that were done leading up to human dosing to ensure that the drug would be safe enough. Toxicology studies performed in cynomolgus macaques at Covance prior to human dosing observed no adverse events even at a 200 mg/kg dose, although neuroinflammatory responses were sometimes observed after chronic dosing in mice. The also cite the literature, reviewed here, arguing that antibodies against certain PrP epitopes may exhibit on-target toxicity [Sonati 2013, Reimann & Sonati 2016], as one reason why they proceeded cautiously in dose escalation).
Why did they dose so high? For one, their preclinical precedent was at a high dose. The mice in [White 2003] got 2 mg twice weekly, which if you assume a 20-30 g animal, is equivalent to 260-400 mg/kg biweekly, more than 2-3 times the dose tested in humans here. Second, the authors indicate that animal pharmacology studies showed that PRN100 was subject to a phenomenon called target-mediated drug disposition (TMDD) [An 2020]: the antibody binds to PrP, which causes it to get internalized and then degraded inside the cell. So perhaps they had to dose high enough to saturate peripheral PrP, in order to leave enough PRN100 in circulation for some to get into the brain. Then there’s the question of how much antibody can get into the brain from circulating blood. An oft-cited statistic is that 0.1% of antibody in blood crosses the blood-brain barrier [Poduslo 1994, Atwal 2011]. However, some of the original data behind that statistic are based on a CSF:serum ratio, which may not always be the relevant metric. Denali Therapeutics found that its BACE1 antibody tested in mice and monkeys had a CSF:serum ratio of ~0.1% but a brain:serum ratio an order of magnitude lower, around 0.01% [Kariolis 2020]. Only by engineering a novel transport vehicle into the antibody were Denali able to get the brain:serum ratio up to around 0.3% - 1%. In the present study, PRN100 exhibited a peak CSF:serum ratio of 0.1% or better across all patients, but brain data are only available from one patient. That patient had brain concentrations of PRN100 estimated at 66-183 nM. On the face of it, this sounds rather encouraging. We estimate that in human brain, PrP is present at roughly 50 - 200 ng per g of brain tissue depending on the region [Mortberg 2022], which (assuming a 35 kDa molecular weight and, again, that 1 g brain ≈ 1 mL volume) corresponds to a concentration of up to 6 nM. That would suggest that PRN100 reached brain concentrations roughly 10-30x the level required to saturate PrP. (The authors also note that in cell culture, ICSM18 had an IC50 of about 1 nM [Antonyuk 2009, Fig S1], implying the concentrations in CSF and brain here were well above the effective concentration, although the expression level of PrP in brain vs. cultured cells may be different, so it is hard to extrapolate). On one hand, the claim that a potentially effective brain concentration of antibody was reached is plausible given the high doses used here. Indeed, TMDD may even promote brain uptake: one study of a BACE1 antibody found that brain concentrations of the drug depended upon expression of BACE1 [Atwal 2011], implying that for highly brain-expressed targets, TMDD could actually drive the drug into brain tissue. On the other hand, there are a few reasons why it’s difficult to be confident that a therapeutically relevant dose was indeed reached where you need it — on the surface of every neuron. Around 1-6% of the volume of a brain is just blood [Copen 2016], so if the drug concentration measured in postmortem brain is mid-nanomolar, it’s hard to be certain that some or even all of this isn’t just driven by a low micromolar concentration in blood. In experimental animals, people get around this problem by perfusing the animals with saline, but in humans, there’s always blood left in postmortem brain. The authors note that in animals, due to TMDD, PRN100 dropped quickly below 1.3 μM in blood, but even that concentration is 20 times the concentration observed in some brain regions here, and so does not exclude a contribution of blood to the signal measured in brain. (The supplement states that brain tissue used for drug concentration measurement was “washed and homogenised with additional washing steps to remove blood and plasma contamination” before analysis, and that this protocol was “was developed and validated in a mouse model in which ICSM18-treated mice were perfused to wash out circulating antibody and compared with non-perfused mice”, but those data are not shown, and it strikes me as rather difficult to remove all blood contamination, particularly since the post-mortem interval was 9 days. I would really need to see some convincing evidence in this regard.) Blood vs. brain aside, the postmortem drug concentration data came from just one patient who died just 3 days post-dose, and so can’t answer the question of whether the dosing regimen here sustained brain parenchyma concentrations expected to saturate PrP continuously over the 2 weeks between doses. In addition, these were bulk tissue measurements, so we can’t infer how deeply the antibody diffused into the brain beyond the vasculature.
Dosing aside, another interesting learning from this study concerns the recruitment and endpoints in a symptomatic prion disease population. In the previous randomized clinical trials of quinacrine and doxycycline in sporadic CJD [Geschwind 2013, Haik & Marcon 2014], patients were diagnosed based on symptomatology combined with EEG or MRI; many were quite impaired at the time of recruitment, with about half either intubated or ventilated by the time of randomization in the doxycycline trial; and overall survival was the only endpoint, without any way of controlling for the disease stage at time of enrollment. The new study, while small, illustrates some of the ways that the advances in the prion field over the past decade could help to empower symptomatic trials in prion disease in the future. All 6 participants were positive for RT-QuIC, a strictly prion disease-specific biomarker. This sort of patient selection biomarker should serve to increase confidence in the diagnosis, and may enable catching patients earlier. All 6 participants retained at least some cognitive and executive function at the time they were enrolled, and 3 were even deemed to still have capacity to consent. Finally, while overall survival was also considered, one analysis in the paper compared the treated patients to historical controls who were matched on disease subtype, starting MRC Scale score, and PRNP genotype. These factors might help to control for some of the variability in survival time.
The authors conclude that the next step should be a formal Phase II clinical trial of PRN100. Of course, my biggest hopes are on PrP-lowering therapy, where the therapeutic hypothesis is the strongest and where we’ve done extensive studies in animal models of its relevance across various disease stages when prion infection has taken hold in the brain [Minikel 2020]. But binding PrP is also a reasonably strong therapeutic hypothesis, and for me, the deeper and more diverse the pipeline of prion disease drug candidates, the better. Will it happen? PRN100 has been on the shelf for 20 years without any big pharma partnering on it, such that when it finally did enter first-in-human, it was an academic-led compassionate use treatment in just 6 people. What has held the industry back from investing in this, and will the new study change that?
Based on my years of interacting with the pharmaceutical industry, I can try to guess at how they’d think about all this. The pivotal question in such a drug development program would be whether a (and in particular, this) PrP monoclonal antibody has a therapeutic window for treatment of prion disease in the brain. This question has never been answered in animal models, nor is it answered by the present study. More granular questions one might ask are: whether the drug can reach meaningful concentrations in the brain, and whether dosing it at such concentrations is safe. The lack of adverse events, even at 6-10 gram doses, is encouraging, but data on drug concentration in brain parenchyma are limited, and there is no target engagement biomarker to convince you empirically that the dose was “enough”. The dose used here is so high that some in industry might call it “undevelopable”, though if you really push on that, it’s not obvious what precludes it. Certainly, prion disease is rapidly fatal enough to justify extreme measures, and this study now rules out a severe toxicity. Some industry folks may simply feel uncomfortable being at the outer edge of clinical precedent in terms of antibody dose level. Other analysts might point to cost. The cost of manufacturing antibodies at scale has been estimated at $100/g [Kelley 2009]. That’s a negligible fraction of what companies charge for approved drugs, but small scale manufacture is surely far more expensive on a per-gram basis (I couldn’t find a good reference) and even at $100/g, if your dose is as high as 10 g every 2 weeks, even this price could actually comprise a non-negligible fraction of the cost of a running a clinical trial. Overall, the present study does a bit to de-risk the prospect of developing PRN100 as a drug, but it leaves the biggest questions unanswered.
Whether or not the new data are enough to launch this long-stalled the drug candidate into Phase II, new technologies could always breathe a new life into PrP monoclonal antibody development. In the decades while PRN100 was in development, there have been new discoveries such as pH-dependent “catch and release” antibodies engineered to avoid TMDD [Igawa 2010], and new ways of hijacking of the transferrin receptor to cross the blood-brain barrier [Kariolis 2020]. Such approaches could eventually allow lower doses of antibody to reach higher concentrations in the brain. Whether that will in turn prove effective, remains to be seen.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.