Reports of small molecule PrPC ligands
Motivation
As I’ve discussed on this blog again and again, the most potent small molecules against prion disease in vivo have all proven strain-specific, extending life in mice infected with some prion strains but not others. The mechanism of action of these molecules — IND24, cpd-b, and anle138b — is not totally clear, but none of them have been suggested to bind PrPC [Kawasaki 2007, Wagner 2013, Ghaemmaghami 2014]. Binding PrPC is an appealing mechanism of action because one might hypothesize that it would stabilize PrPC and prevent its conversion to any prion strain. In support of this, there have been several proofs of concept that monoclonal antibodies against PrPC can block prion propagation and/or neurotoxicity in various model systems [Peretz 2001, White 2003, Sonati 2013], though there is heated debate about which epitopes can be safely targeted [Solforosi 2004, Klohn 2012, Sonati 2013, Herrmann & Sonati 2015].
Binding and stabilizing the native state of an otherwise amyloidogenic protein is the mechanism of action of tafamidis, which, as far as I know, is the only truly disease-modifying drug approved in any country for any amyloid disease. But tafamidis binds to a pocket on transthyretin that evolved to bind thyroid hormone [Bulawa 2012]. Finding small molecules that bind PrP — or any other protein that lacks a binding pocket speciically evolved to bind small molecules — might be a more difficult task.
While there are well-established methods, of various throughput levels, for identifying small molecules that bind to a protein — thermal shift [Pantoliano 2001], circular dichroism, surface plasmon resonance, small molecule microarray [MacBeath 1999] and so on — there are also well-established sources of false positives in binding assays. One of the most insidious such sources is that some small molecules will form aggregates, and the aggregates will then bind to a protein, resulting in promiscuous non-specific binding at high enough concentrations [McGovern 2002].
Despite the obvious challenges in identifying a small molecule that specifically and safely binds PrPC, the approach retains enough conceptual appeal that I decided I should educate myself on what is known so far. This post is therefore devoted to exploring the progress to date in binding PrPC with a small molecule. What approaches have been attempted? What pitfalls have been identified? What molecules have been claimed to exert a therapeutic effect by binding PrPC, and are any of the claims credible?
Are there known molecules that bind PrPC?
When I looked in the literature, I found a large number of claims asserting that various molecules bind PrPC. One challenge in interpreting these claims is that almost anything will bind almost anything if the concentrations are high enough. If one looks for a molecule that binds PrPC at a concentration comparable to the concentration at which it is bioactive against prion formation, then the field of candidates grows much narrower.
One group has published NMR data showing an interaction of quinacrine with the C terminus of PrPC [Vogtherr 2003], but those experiments were done at 10,000x the cellular EC50 for quinacrine, calling into question whether this binding event could be responsible for quinacrine’s activity in cell cultures. And as reviewed in this post there are many reasons to suspect that quinacrine’s relevant mechanism of action in prion-infected cells may be something else. Similarly, there are a crystal structure and NMR data supporting a direct interaction of PrPC with promazine and chlorpromazine [Baral 2014], but again, very high concentrations were used: the PrP crystals were soaked in 1-10 mM chlorpromazine solutions, and the NMR was done at 0.5 to 12 mM chlorpromazine, in contrast to a reported cellular EC50 of just 3 μM [Korth 2001]. Again, there are also plenty of other proposals for chlorpromazine’s mechanism of action with respect to prions [Yamasaki 2014]. Also, the Collinge lab found no evidence that either of these binds PrPC at 100 μM [Nicoll 2010].
Then there is Congo red, which is well-known to inhibit the accumulation of PK-resistant PrPSc in cell culture, with an EC50 somewhere below 140 nM [Caughey & Race 1992]. It has twice been reported to yield a signal in surface plasmon resonance with PrPC [Kawatake 2006], [Touil 2006]. The signal was far greater than that observed for quinacrine, but still, the KD of Congo red for PrPC was estimated at 1.6 μM [Kawatake 2006], at least an order of magnitude higher than the cellular EC50. Congo red retains its activity in cell-free solid state prion conversion reaction [Demaimay 1998], hinting that it interacts directly with some conformation of PrP, rather than an indirect cellular target. Congo red is an amyloid-binding dye, indeed, it is tautologically so, as one of the tinctoral definitions of amyloid is that it binds Congo red [Fowler 2007]. Given that this compound binds the amyloid conformation of diverse substrate proteins, it seems parsimonious to suppose that its antiprion activity is mediated by binding to PrPSc rather than to PrPC. In support of this, its EC50 in the cell-free reaction is more dependent on the concentration of PrPSc than on that of PrPC [Demaimay 1998], though as the authors point out, interpretation of this is complicated by the fact that PrPSc is present in molar excess in that particular reaction. It would be interesting to repeat this experiment using the modern cell-free paradigms of RT-QuIC or PMCA, in which the substrate is present in molar excess.
Moving along, there are also the sulfonated, and usually metallated, anionic porphyrins and phthalocyanines, the most potent of which have cellular EC50s at or below 1 μM [Caughey 1998] and can delay onset of disease by >50% in peripherally prion-infected mice [Priola 2000, Priola 2003]. Of course, these are charged molecules and don’t cross the blood-brain barrier. When injected directly into the brains of intracerebrally infected mice, they had only marginal effects on the disease course [Kocisko 2006a]. Some of these molecules have been shown to be active in a solid state cell-free conversion assay [Caughey 1998]. Phthalocyanine tetrasulfonate (PcTS), like Congo red, yields a signal indicating PrPC binding in surface plasmon resonance, but with a KD estimated at ~18 μM [Kawatake 2006, Dee 2012], about 1.5 orders of magnitude higher than its cellular EC50 of 0.5 μM [Caughey 1998]. The stoichiometry of this binding event was estimated to involve 4 or 5 PcTS molecules binding to each PrPC molecule for truncated PrP (SHaPrP 90-232), with an additional ~5 binding sites in the unstructured N-terminal tail of PrP (SHaPrP 29-90) [Dee 2012]. One study found that PcTS and the anionic porphyrins do bind PrP, but form an aggregate or colloid, rather than interacting with PrP in a simple 1:1 manner [Nicoll 2010]. Some of these molecules are fairly large, and by the time we get to PcTS, we are no longer talking about a single chemical entity - it is in fact a mixture of isomers with 3 or 4 sulfonate groups in different positions [Caughey 1998, see legend of Figure 1].
One in silico screen uncovered a molecule, dubbed GN8, which was predicted to bind PrPC and subsequently shown to reduce PK-resistant PrPSc accumulation in cell cultures, with some evidence for a bit of in vivo activity as well [Kuwata 2007], but both the Collinge and Harris labs have been unable to replicate the original group’s finding that this molecule actually binds PrPC [Nicoll 2010, U.S. Patent Application WO2014025785A2].
The most recent claim of a small molecule binding PrPC has been Chicago sky blue 6B [Risse 2015]. It was estimated to bind PrPC with a 3:1 stoichiometry and to have a KD of 0.55 μM (by isothermal titration calorimetry) and a cellular EC50 of ~3 μM (just eyeballing Fig 7A) for clearing PK-resistant PrPSc from PK1 cells. While still almost a 10-fold difference, this is the closest match so far between a compound’s affinity for PrPC and its bioactive concentration. The compound was observed to bind full-length PrP but not PrP 119-231, and it occludes Aβ oligomer binding, which has been mapped to residues 23-31 and 95-110 [Fluharty 2013]. These two regions each contain a cluster of positive charges, and Chicago sky blue has four formal negative charges, so I would speculate that electrostatics may be involved in this interaction. While this compound may bind PrPC, it does not do so at a single defined binding site, and it was toxic at a concentration barely higher than its EC50, so it is probably not a promising lead for future development.

Above: a selection of small molecules that have been reported to bind PrPC, but in a multivalent fashion and/or at concentrations far above the concentration required for activity in cells.
For at least one of the porphyrins, though, there is rather compelling evidence for binding PrPC, at a defined site, in a simple 1:1 binding mode [Nicoll 2010]. A cationic porphyrin called FeTMPyP (originally referred to as T(N-Me-4-Py)P-Fe3+ [Caughey 1998, Figure 4]) was shown by several different methods — equilibrium dialysis, isothermal titration calorimetry, circular dichroism, sedimentation velocity analytical ultracentrifugation to bind HuPrP [Nicoll 2010]. The various methods employed were in pretty good agreement on the affinity, with a KD ranging from 3.2 to 8.1 μM, depending on the exact conditions, measurement method, and whether HuPrP 91-231 or 119-231 was used. NMR gave evidence for specific binding to residues ~160-180. According to circular dichroism, the compound raised the melting point of HuPrP 91-231 from 66.1 to 68.4°C when the compound was present at 18 μM and the PrP was present at 6.5 μM, which is almost a 3:1 molar ratio. It would be interesting to know if this result can also be obtained in a 1:1 stoichiometry. But in prion-infected PK1 cells, at least, it reduced PK-resistant PrPSc at an EC50 of 1.6 μM. This is by far the best match between a compound’s affinity and its effective concentration, and together with the NMR data and the lack of any observed aggregation, seems to make it reasonably likely that this molecule really does bind PrPC in a simple 1:1 fashion.

Above: FeTMPyP, the molecule with the strongest evidence for having antiprion properties mediated by binding PrPC in a 1:1 fashion.
It is also worth discussing another class of reported PrPC binders, which are large, heterogeneous, anionic, usually sulfated polymers: some of the best-studied examples include pentosan polysulfate, heparin, dextran sulfate 500, and phosphorothioated oligonucleotides. Many of these compounds reduce PK-resistant PrPSc in ScN2a cells [Caughey & Raymond 1993, Kocisko 2006b, Karpuj 2007], and pentosan polysulfate is sometimes even used as a tool compound to cure cells of prion infection [e.g. Bate 2004]. Some of them considerably slow down peripherally acquired prion disease in mice [Ehlers & Diringer 1984, Diringer & Ehlers 1991, Farquhar 1999], and pentosan polysulfate even works against intracerebrally acquired prion infections [Doh-Ura 2004], though it is problematic in terms of delivery, timing, and complications, which may explain its lack of success in human trials [Bone 2008, Tsuboi 2009].

Above: the monomeric subunits of large, anionic polymers that are reported to bind PrP, inhibit PK-resistant PrPSc accumulation in cells, and/or extend survival in animals infected with prions.
Do these polymers work by binding PrPC? The evidence is mixed. Heparin conjugated to beads has been observed to pull down PrPC [Caughey 1994], pentosan polysulfate has been observed to bind PrPC according to surface plasmon resonance [Brimacombe 1999], and heparin and other glycosaminoglycans have been observed to bind PrPC in ELISA and flow cytometry [Pan 2002]. Pentosan polysulfate and dextran sulfate (as well as Congo red) have been reported to induce endocytosis of PrPC in uninfected cells [Shyng 1995]. Heparan sulfate and pentosan polysulfate have been reported to increase the rate of cell-free prion conversion in a solid-state assay [Wong 2001], which is consistent with binding to either PrPC or PrPSc, though it is surprising in light of the antiprion properties of these compounds in cell culture systems. Endogenous glycosaminoglycans similar to these compounds have long been known to be incorporated into prion plaques in the brain [Snow 1989], and seem to play a role in binding PrPSc in cell culture experiments as well [Hijazi 2005]. There is a vast literature on the interaction between glycosaminoglycans, and these polyanions, and PrP, and I’ve only just touched on it here.
In a recent review, Emiliano Biasini speculates that these polyanions may simply act by non-specific electrostatic binding: they have lots of negative charges, and PrP’s N terminus is highly flexible and has lots of positive charges [Iraci 2014]. That explanation makes sense, but if it’s true, then some of these polyanions exhibit a shockingly strong non-specific interaction. Consider the stoichiometry involved. Because the molecules shown above are large polymers of variable molecular weight and sometimes variable branching, most of the studies simply express their concentration in terms of ng/mL or similar rather than molarity. Pentosan polysulfate has an EC50 of only about 1 ng/mL [Caughey & Raymond 1993, see Figure 6], and its monomeric unit (shown above) has a molecular weight of about 320 g/mol. By my calculations, means that to reduce PK-resistant PrPSc accumulation in cells by 50%, all you need is pentosan polysulfate polymers containing the equivalent of (1e-6 g/L)/(320 g/mol) = 3 nM worth of monomers. This would make pentosan polysulfate, in a sense, the most potent known antiprion compound.
Whatever the mechanism of action of the polyanions, they have only been reported to have activity when used as polymers, and even though a couple of them (pentosan polysulfate and heparin) are actually approved drugs, they are certainly not “drug-like”, there seems little prospect of identifying a specific binding site on PrP, and they have no hope of crossing the blood-brain barrier. So these molecules, too, are not really the PrPC binder that we are looking for.
I don’t believe that my coverage of the literature has been completely exhaustive (and feel free to let me know about additional claims I’ve missed), but at this point I believe I’ve touched on many of the more thoroughly investigated molecules reported to bind PrPC. I next turned to searching the patent literature. The search results are dominated by peptide or antibody ligands [e.g. US 8605161 B2, US 20140294844 A1], and by several versions of a patent application by Prometic Biosciences Limited, covering the use of substituted triazines [US 8030484 B2]. Although those are small molecules, none of the various versions of that patent appear to contain any real data demonstrating that such compounds actually bind PrPC.
The most interesting patent I found was an application published last February, filed by David Harris, Emiliano Biasini, and the Trustees of Boston University among others [WO2014025785A2], covering a series of small molecules shown to be active in any or all of the following assays:
- Raising the melting temperature of recombinant mouse PrP.
- Inhibiting PK-resistant PrPSc accumulation in ScN2a cells.
- Preventing Aβ oligomers from binding PrPC in cell-free experiments.
- Preventing spontaneous transmembrane currents in cells expressing PrP ΔCR. †
- Binding to recombinant mouse or human PrP according to surface plasmon resonance.
†For background on PrP ΔCR see [Solomon 2010]
I did not find a publication on PubMed or Google Scholar covering this work, so it is likely that the experiments in the Harris lab are ongoing, and the patent application, which has a priority date in 2012, probably represents a rather outdated snapshot of the current science on these compounds. Nevertheless, it was an interesting read. The compound for which the largest amount of data is discussed in the patent is one dubbed DS26, which is sometimes referred to in the text as 2-(2R,3S,5R,6S-pentahydroxycyclohexylidene) hydrazine carbothioamide. According to the IUPAC-parsing tool OPSIN [Lowe 2011] this doesn’t quite parse, so I asked my colleague Zarko Boskovic, who pointed out that on line 00348, describing its synthesis, it is referred to by its full IUPAC name, 2-((2R,3R,4S,5R,6S)-2,3,4,5,6-pentahydroxycyclohexylidene)hydrazinecarbothioamide, which looks like this (only the E enantiomer is active according to line 00398):

According to in silico docking, the molecule is predicted to bind what they call “PDB-1”, a set of residues defined in line 00380. I have highlighted these residues in teal in PyMOL on MRC Prion Unit’s human PrPC structure [PDB# 2W9E, Antonyuk 2009]:
bg_color white
fetch 2w9e
hide everything
show surface, chain A
color yellow
color 0x36648B, chain A and (resi 134+135+136+137+150+154+155+157+158+159+160+209+210+213)

Above: the predicted binding site of DS26 on the surface of PrPC according to in silico docking.
I was surprised to see that the predicted binding site is apparently a ridge on the surface of the protein, rather than a valley.
The inventors used surface plasmon resonance to estimate a KD of 800 nM (see line 00389) for DS26 binding to human PrPC. They found that it increased the melting temperature (Tm) from 66.8 to 68.9°C (line 00406). They also say they tested its ability to inhibit PK-resistant PrPSc accumulation in ScN2a cells. The EC50 is not given, but is stated to be “considerably higher” than the KD (line 00408), and the lowest concentration they report having tested in cells was 10 μM. The compound did, however, exhibit nearly complete inhibition of transmembrane currents in PrP ΔCR-expressing cells at a slightly lower concentration, 5 μM (line 00410).
So is this finally the molecule that has been so elusive — a small molecule that binds PrPC in a 1:1 stoichiometry at a defined binding site? It is surely too early to judge based on just this patent application. Certainly, DS26 is not subject to the same objection as quinacrine and chlorpromazine. Those two molecules have been observed to bind PrPC, but only at concentrations far too high to explain their very low EC50s in cells. DS26, if anything, is the opposite: it binds PrPC at only 800 nM, yet doesn’t achieve activity against PrP ΔCR until closer to 5 μM and against PrPSc until at least 10 μM. This is not as close of a match between affinity and bioactivity as was reported for FeTMPyP (see above).
This is just a lead compound, and in its current form, at least, it is probably a long ways from being a drug. According to ChemDraw, this molecule has predicted logP of -3.49, meaning it’s quite hydrophilic. For comparison, I used rcdk to predict the XlogP for all 221 FDA-approved CNS drugs. DS26 is more hydrophilic than any FDA-approved CNS drug:
require(rcdk)
drugs = read.table('http://www.cureffi.org/wp-content/uploads/2013/10/drugs.txt',header=T,sep='\t',quote='',comment.char='')
cns = parse.smiles(drugs$smiles[drugs$cns_drug])
xlogp = eval.desc(cns, "org.openscience.cdk.qsar.descriptors.molecular.XLogPDescriptor")
range(xlogp$XLogP)
hist(xlogp$XLogP,breaks=(-10:16)/2,col='#FF9912',border=NA,axes=F,main='Predicted LogP',yaxs='i',xaxs='i',ylab='',xlab='')
abline(h=0)
axis(side=1,at=-4:7,lwd=0,lwd.ticks=1)
axis(side=2,at=(0:3)*10,lwd=0,lwd.ticks=1,las=2)
mtext(side=1,text='\u2190more hydrophilic more hydrophobic\u2192',line=2)
mtext(side=1,text='Predicted logP',line=3,font=2)
mtext(side=2,text='Number of compounds',line=2)
rect(xleft=-3.74,xright=-3.24,ybottom=0,ytop=1,col='#D91309',border=NA)
arrows(x0=-3.49, x1=-3.49, y0=5, y1=1.5, length=.05, angle=30, code=2, col='#D91309')
text(x=-3.49,y=5,labels='DS26',col='#D91309',pos=3,cex=.9)
arrows(x0=-1, x1=0, y0=20, y1=20, length=.05, angle=30, code=2, col='#FF9912')
text(x=-1,y=20,pos=2,labels='CNS drugs',col='#FF9912',cex=.9)
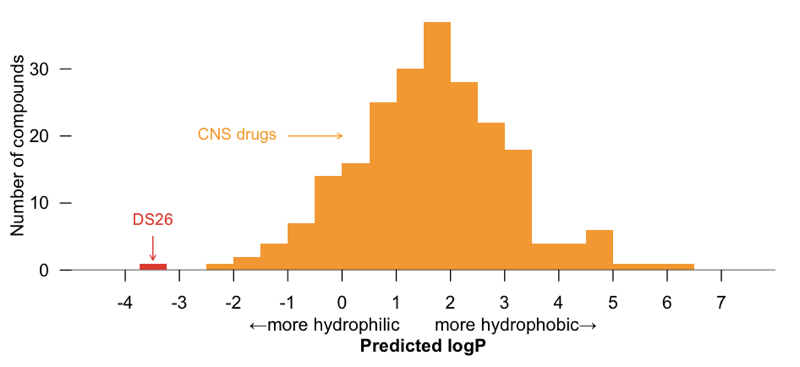
But those are drugs, and this is still just a lead; perhaps it could be pushed towards BBB permeability through some medicinal chemistry optimization or a prodrug approach [reviewed in Rautio 2008a, Rautio 2008b]. Indeed, many of the analogues discussed in the patent contain additional hydrophobic groups substituted in at the NH2 at right. But whether or not an analogue of DS26 ever becomes a drug, if it really does bind PrPC, in a monovalent fashion and at a specific binding site, it could certainly prove to be a useful tool compound. I look forward to watching this story develop.
Sadly absent from my patent search results was any news from London. MRC Prion Unit’s website has for years contained a page describing joint efforts with GlaxoSmithKline to find small molecules that bind and stabilize PrPC, and they wrote a review arguing for binding PrPC as a small molecule strategy [Nicoll & Collinge 2009] but so far there has been no word on the results of these efforts. John Collinge presented survival curves from mice treated with two compounds dubbed GSK01 and GSK02 at last year’s CJD Conference, but he said that those particular compounds “have a unique mechanism of action and do not bind PrP”. Whether the screen has turned up any leads that do bind PrP, I am not sure — as far as I can tell, no patent has been published yet.
This may be all we have so far, then. While there are some molecules, large and small, that appear to interact directly with PrP, FeTMPyP [Nicoll 2010] appears to be the only one with pretty solid evidence for binding PrPC at a single, well-defined site at a therapeutically relevant concentration. If you the reader know of any breakthroughs I’ve missed, please leave a comment below.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.