The N-terminal toxicity hypothesis
After they had shown that PrP knockout mice were resistant to prion disease [Bueler 1993], Charles Weissmann and Adriano Aguzzi set out to check whether PrP transgenes would rescue the susceptibility to prion infection, and found that they did [Fischer 1996]. Because PrPSc was operationally defined based on its resistance to limited protease digestion, with proteinase K only clipping off the N terminus up to residue 80, they also wanted to check whether PrP lacking the N terminus up to residue 80 could likewise rescue susceptibility. Of course, they left the signal peptide, resiudes 1-22, intact, so that the protein would still be co-translationally translocated into the ER. They weren’t certain whether this localization would still occur correctly if they also deleted the residues right after the signal peptide, so they also left residues 23-31 intact, and created mice expressing PrPΔ32-80 [Fischer 1996]. These mice were susceptible to prions, and thus began a sort of binary search to see how much of the N terminus could be deleted without rendering PrP incapable of prion propagation.
The answer eventually turned out to be somewhere around residues 93 and 106 in mouse numbering [Aguzzi 2008]. But mice expressing the first two deletion mutants they tested, Δ32-121 and Δ32-134, had a different problem: they suffered a spontaneous neurodegeneration of the cerebellum, which was not transmissible, didn’t involve PrP aggregation, and occurred only on a knockout background (it could be rescued by co-expression of full-length PrP) [Shmerling 1998]. The original decision to leave residues 23-31 intact in the so-called “Shmerling mice” proved to be serendipitous. Stanley Prusiner and David Harris each later created other mutants deleted all the way out to residue 23, and these all proved to be benign (see this post for a full review).
So there was something uniquely pathogenic about expressing only a PrP molecule with the N terminal tip present but some part of the middle of the protein deleted. Adriano Aguzzi, and later David Harris, created a series of additional mutants to narrow down the region responsible (again, the full battery of mutants is catalogued in this post). The most severe phenotype to date has been found in David Harris’s ΔCR (PrPΔ105-125) construct — mice expressing this protein suffer neonatal lethality if full-length PrP is not co-expressed [Li 2007].
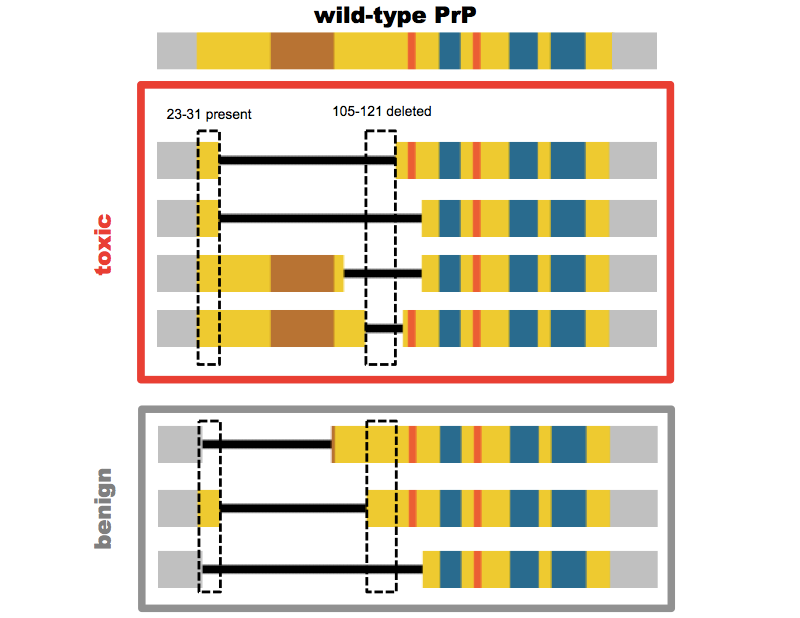
Above: PrP mutants lacking at least residues 105-121 (mouse numbering) but retaining at least residues 23-31 are toxic.
Were the Shmerling and ΔCR constructs just a scientific accident of no relevance to actual prion disease, or were they a crucial toehold into understanding how prions kill neurons? On one hand, there are several different ways that tinkering with PrP can prove toxic (this post talks about a few of them), so perhaps we shouldn’t read too much into any one of them. On the other hand, the idea of a cell surface-localized, non-aggregated toxic species of PrP seemed a conceptually appealing model, given the fact that individual neurons must express PrP in order to be susceptible to prion neurotoxicity [Brandner 1996]. Perhaps full-length PrPC transduced a toxic signal upon contact with PrPSc, and mutants like ΔCR just constitutively transduced that signal.
Moreover, there was an appealing analogy to genetic prion disease caused by octapeptide repeat insertions (OPRI) in PrP’s N terminus. Obviously, those mutations make PrP longer, not shorter. Yet they represent a rather unusual subtype of genetic prion disease. Some cases have been transmitted to primates or mice, but other cases have proven difficult or impossible to transmit [Brown 1994, Mead 2006], and a mouse model with a 9 octapeptide repeat insertion developed spontaneous disease that was not transmissible [Chiesa 1998]. And while prion diseases are usually rapidly progressive dementias with a typical duration of 6 months [Pocchiari 2004], OPRI patients often have disease durations on the order of a decade [Mead 2006]. The age of onset is also heavily dependent on the codon 129 residue of the trans allele, whereas age of onset in, say, E200K or D178N genetic prion disease exhibits little or no dependence on trans codon 129 [Mead 2006, Minikel 2014]. These observations might suggest that something other than (or additional to) the usual prion pathology is afoot in OPRI disease.
And then there are the toxic antibodies. Two years ago, Adriano Aguzzi showed that a series of antibodies against the C terminus of PrP are toxic, both in cerebellar organotypic cultured slices and in vivo, and that antibodies against the N terminus prevent that toxicity [Sonati 2013]. Thus, the toxicity of C-terminal antibodies must be mediated by the N terminus.
So here we have three different paradigms in which the N terminus of PrP seems to be responsible for a toxic signal. The question is, is this just a coincidence, or is the toxic mechanism in each of these instances the same, and more importantly, is it the same toxic mechanism as in bona fide prion infection?
What I’ll call the N terminal toxicity hypothesis holds that yes, in all of these paradigms, PrP is toxic by the same mechanism. Thus, toxic antibodies and interstitial deletions are models of bona fide prion neurotoxicity, and those minimally transmissible OPRI cases are genuine prion diseases, they just present a higher ratio of PrPC’s intrinsic toxicity to PrPSc-induced toxicity.
To be clear, I’m not taking any credit for this hypothesis — nothing here is original thinking on my part. The hypothesis, in the wording that I’ve just stated it here, is sort of an amalgam of different statements variously put forth by Adriano Aguzzi, David Harris, and Glenn Millhauser. For instance:
- “positing that PrPC mediates a neurotoxic signal triggered from the extracellular milieu by PrPSc… The notion that PrPC can transmit toxic signaling is supported by the finding that antibodies binding to PrPC can induce robust neurotoxicity and that mice expressing mutant forms of PrPC that lack certain domains develop spontaneous neurodegeneration” [Aguzzi & Falsig 2012].
- “PrP molecules missing certain critical regions produce dramatic neurodegenerative phenotypes when expressed in transgenic mice, which may reflect alterations in the normal function of PrPC. It remains to be determined whether the same pathways are affected by infectious PrPSc.” [Biasini 2012]
- “Might GD ligands be toxic by emulating the docking of PrPSc to PrPC?… Perhaps PrPSc oligomers engage PrPC through its α1/α3 helices in a similar way to the GD ligands… If the disease caused by supernumerary OR is mechanistically equivalent to that elicited by GD ligands, agents effective against GD ligand toxicity may also be beneficial to patients with OR insertions.” [Sonati 2013]
- “Could this model… help explain other experiments demonstrating neurotoxic effects and perhaps certain aspects of neurodegenerative disease?” [McDonald & Millhauser 2014].
What could be the common denominator between these different toxic paradigms? All of the investigators quoted above seem to agree that the N terminus is what mediates toxicity. It might kill cells by directly forming pores in the membrane, or via an interaction with other proteins such as ion channels [Solomon 2010, and see discussion in Aguzzi & Falsig 2012]. Glenn Millhauser has hypothesized, and has generated in vitro data in support of the notion, that the N terminus normally docks on the face of PrP’s C terminus [Spevacek 2013]. Here’s a whimsical cartoon diagram:
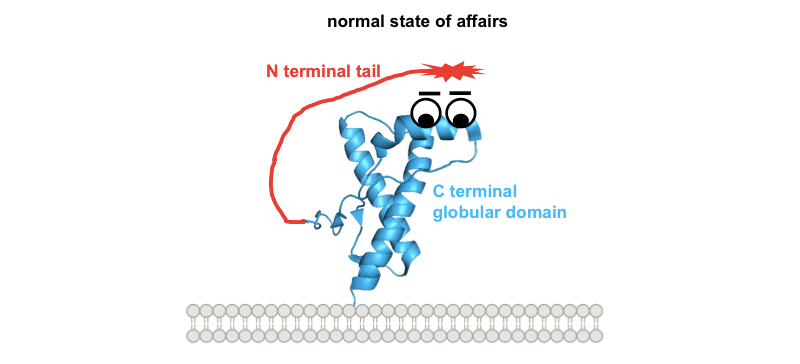
So one possible way to visualize the commonality between these toxic paradigms would be if all four of them prevent this docking in one way or another, freeing up the N terminus to do things it shouldn’t. Building on our earlier cartoon, and for the sake of illustration assuming that the toxic effect of the N terminus is indeed mediated by direct interaction with the plasma membrane:
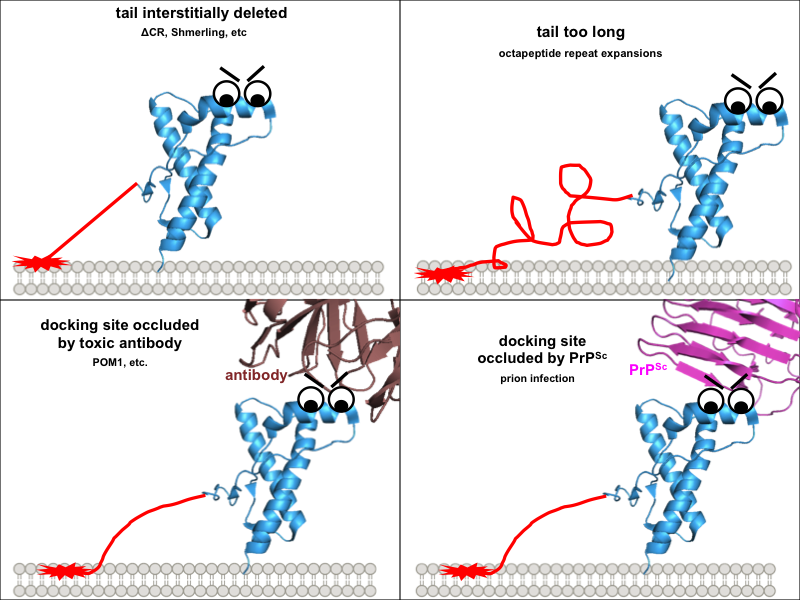
These diagrams might be overly cutesy and simplistic, but the exercise of drawing them forced me to think through several implications of this model. Why are the interstitial deletions toxic? It’s not just that the tail is too short to reach the docking site, because Δ105-125 is actually worse than Δ32-134. Instead it must instead be that the tail is rendered inflexible in some way, or that without its cryptic transmembrane domain it cannot traverse the membrane, or simply that, as Glenn Millhauser has proposed, it is incapable of undergoing alpha cleavage to shut off its toxic activities [McDonald 2014, McDonald & Millhauser 2014]. Speaking of which, these diagrams help to visualize how, in all of these paradigms, alpha cleavage — cutting PrP at residue 111 — would be protective:
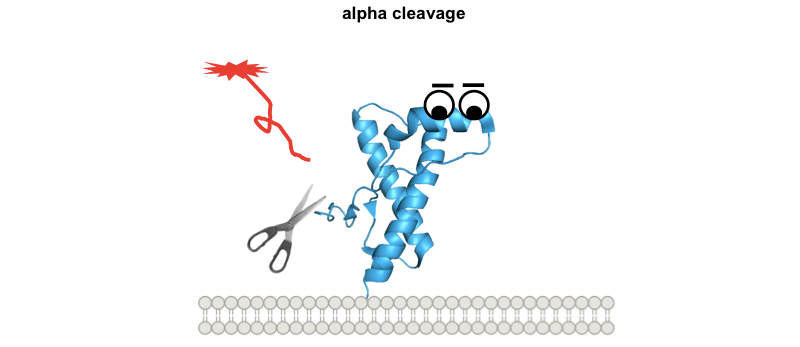
So how does one test the N-terminal toxicity hypothesis? How, in particular, would one show that the toxicity of prion infection operates by the same mechanism as any of these other paradigms? A paper from the Aguzzi lab out earlier this year seeks to show that at least the toxic antibodies and genuine prions do share the same mechanism [Herrmann & Sonati 2015]. They expose cerebellar organotypic cultured slices (COCS) to prions or the toxic POM1 antibody and then test a battery of different genetic manipulations, antibody and tool compound treatments to see if they have the same effect on both models.
There’s a line that I really like from the classic paper showing that PrP knockout mice are resistant to prions:
if [mice devoid of PrP] succumb to the disease or propagate infectivity, albeit without showing symptoms of neurological disease, the protein only hypothesis would be falsified.
— [Bueler 1993]
When possible, I think it’s useful to state up front what the interpretation of each possible result of an experiment would be. This way you can show that a hypothesis is falsifiable, and that it’s not simply the case that just any result could somehow be interpreted in favor of the hypothesis.
So how, if at all, could the N-terminal toxicity hypothesis be falsified, if it were false? As stated above, the experiments in this paper [Herrmann & Sonati 2015] revolve around looking at what does, or doesn’t, rescue COCS from the toxicity of prions, or POM1. That gives a 2×2 matrix for four possible outcomes: yes/no × prion/POM1. The fact that calpain inhibitors, ROS scavengers, and antibodies against PrP’s N terminus all rescue both POM1- and prion-exposed slices from neurodegeneration is interpreted to mean these things block a shared neurotoxic pathway. The fact that pentosan polysulfate, Congo red, and amphotericin B block prion-induced but not POM1-induced neurotoxicity is interpreted to mean they block prion replication, which occurs upstream of toxicity and is not needed for POM1 toxicity. That’s eminently reasonable, particularly in light of the evidence that those treatments resulted in reduced prion titers. The fact that antagonizing glutamate receptors or ER calcium channels didn’t block prion- nor POM1-induced damage is certainly a useful observation, given the prior literature suggesting excitotoxicity and glutamate signaling [You 2012, Watt 2012, Um 2013] play a role in PrP toxicity. But in my view, this doesn’t particularly provide evidence for or against the N-terminal toxicity hypothesis. If you screened a library of hundreds of thousands of compounds for ones that block prion- or POM1-induced toxicity, you would presumably find, just as for any other cellular phenotype you cared to screen against, that the overwhelming majority of compounds are inactive. This doesn’t tell you much, because even if prion- and POM1-induced neurodegeneration had nothing at all in common, it would still be the case that >99% of compounds fail to influence either phenotype.
This leaves one last cell in the 2×2 matrix: what if a particular treatment rescued POM1 neurotoxicity but not prion neurotoxicity? Perhaps this result is the one that would falsify the N-terminal toxicity hypothesis:
| rescues prion-infected slices | rescues POM1-exposed slices | interpretation |
|---|---|---|
| yes | yes | blocks a shared neurotoxic pathway |
| yes | no | blocks prion replication, upstream of neurotoxicity |
| no | yes | would falsify the N-terminal toxicity hypothesis?? |
| no | no | none |
And yet isn’t there already one experiment that has given exactly this result? In Figure 3A of [Sonati 2013] it is shown that COCS expressing PrPΔ32-93 are resistant to POM1 toxicity. Yet Δ23-93 mice are still susceptible to prion infection [Flechsig 2000]. Does this falsify the hypothesis? Well, not quite. Though Δ32-93 mice can be infected with RML prions, and eventually do succumb to lethal disease, they have much longer incubation times than mice expressing comparable levels of wild-type PrP [Flechsig 2000]. At first glance, this would seem to mesh perfectly with the N-terminal toxicity hypothesis: deletion of 32-93 abolishes N-terminal toxicity, so that prions need to replicate longer, to reach higher levels, before orthogonal toxic pathways finally overwhelm neurons. But if that were the case, then prion titers in terminally ill Δ32-93 mice should be higher than in mice expressing wild-type PrP, whereas in fact they’re reported to be 1.5 logs lower. This is true regardless of whether the indicator mice used for titer determination express wild-type or Δ32-93 PrP, indicating it’s not merely an issue of heterologous PrP molecules confounding titer calculations. [Flechsig 2000]. In addition, the pathology in the Δ32-93 mice is atypical, with little neuronal loss in the brain. For all these reasons, I think it’s hard to know what to make of the data.
If one wanted to set out to falsify the N-terminal toxicity hypothesis, other PrP mutants might provide a more interpretable result. Mice expressing PrP Δ32-80 are fully susceptible to prions, with typical pathology and with incubation times no different from mice expressing wild-type PrP at comparable levels [Fischer 1996]. So if COCS expressing Δ32-80 were to prove resistant to POM1 toxicity, then that would, if not completely falsify the hypothesis, at least show that N-terminal toxicity is not a major determinant of the tempo of bona fide prion disease in vivo. A separate attempt to falsify the hypothesis might use N-terminal deletions that extend all the way to the N terminus. Mice expressing PrP Δ23-88 are perhaps not a perfect model here, since when inoculated with prions they develop typical prion pathology and scrapie signs but over quite protracted incubation times [Supattapone 2001]. A better choice might be Δ23-88, Δ141-176 or “miniprion” mice [Supattapone 1999]. At least on second passage, these mice are highly susceptible to RML prions, with rapid incubation times and typical prion neuropathology, so again, if COCS prepared from these mice were fully resistant to POM1 toxicity, that would invalidate the N-terminal toxicity hypothesis.
Indeed, the mere fact that mice with so much of the N terminus deleted can still become prion-sick raises some questions. When I first saw Adriano Aguzzi present some of these data, at the Whitehead Institute last year, I asked how his theory of N-terminal toxicity might mesh with the existence of such mice. He responded that there simply must be more than one neurotoxic mechanism. That’s quite reasonable, and it’s important to recognize that these things needn’t be mutually exclusive.
Perhaps the question, then, is how much of the damage caused by prions can be attributed to the putative toxicity of the N terminus. The Aguzzi lab’s battery of tool compounds has pointed to three different pathways that appear to lie downstream of N-terminal toxicity: calpain activation, ROS production, and PERK phosphorylation. For at least the latter two, various experiments have addressed the degree to which each of these determines time to terminal illness in vivo. Inhibition of ROS production by knockout of Nox2 is reported to extend survival by +4% [Sorce 2014]. Overexpression of human SOD1 is also reported to extend survival by +19% [Tamguney 2008], though SOD1 is cytosolic and the Aguzzi lab has found that the operative ROS species mediating prion and POM1 toxicity are extracellular [Herrmann & Sonati 2015]. Inhibition of PERK activation by lentiviral overexpression of GADD34 [Moreno 2012] or with a small molecule PERK inhibitor [Moreno 2013] seems to extend survival by something like 12%. To my knowledge, calpain inhibition has not been tested as a therapeutic strategy in vivo. (Confusingly, one calpain inhibitor, known as E64d or loxistatin, was originally reported to inhibit PK-resistant PrPSc formation with an EC50 of 500 nM in neuroblastoma cells [Doh-Ura 2000], but was later reported to rescue COCS from prion neurotoxicity while actually allowing increased PK-resistant PrPSc [Falsig 2012]. This compound is apparently tolerated in vivo, as it went to human clinical trials for muscular dystrophy, where it failed on efficacy, not safety [Satoyoshi 1992]. But I didn’t see any studies where it was used to treat prion-infected mice.)
But at least from what’s been reported so far, all of these downstream neurotoxic pathways seem to add up to a pretty small quantitative impact on the disease course, something like 4% + 12%. This isn’t anywhere near the 2- or 4-fold increase in survival time that has been achieved with small molecules targeting prion propagation. There are many possible explanations for this — maybe N-terminal toxicity is a major determinant of neurodegeneration, and it’s just that calpains, ROS, and PERK all put together only mediate a fraction of the damage wrought by the N terminus. If so, then going further upstream — say, by engineering mice that constitutively express antibodies against PrP’s N terminus in their brain — should achieve much greater neuroprotection than any of these tool compounds. An alternative is that maybe the N terminus only causes marginal neuronal damage compared to some larger, orthogonal toxic mechanism that requires PrP expression but does not specifically require the N terminus to be present.
And as long as we’re talking about alternatives, obviously, the N-terminal toxicity hypothesis as I’ve stated it here is rather sweeping; some subset of the toxic paradigms diagrammed above could be equivalent to each other without all four being equivalent. For instance, maybe it could turn out that interstitial deletions and toxic antibodies are exquisite models of the toxicity observed in OPRI patients, but do not reflect the dominant neurotoxic pathways of prion infection in vivo.
But no matter what, it seems, we will be stuck with either one of two unsatisfying and seemingly unparsimonious situations. If the hypothesis is true, then we must reconcile the universality of N-terminal toxicity with the fact that mice lacking the entire N terminus can still suffer a rapid, lethal, and apparently typical-seeming prion infection. If the hypothesis is false, then we have to simply accept as coincidence the uncanny similarities between these paradigms, such as their PrP-dependent toxicity to neurons, and their abrogation by antibodies against PrP’s N terminus and by the other treatments discussed above. Perhaps there is a third possibility — I would love to hear it.
Regardless of whether N-terminal toxicity is identical to bona fide prion toxicity, and whether the real mechanism looks anything like the cartoons in this post, the data certainly show that the toxicity is real. Do the antibodies really just sterically exclude the N terminus from docking, or do they induce a larger conformational change? Does the N terminus really interact with the membrane? Is it just a coincidence that the deleted region is a cryptic transmembrane domain, or does PrP normally pass through the membrane from time to time, and are the deletion mutants toxic because they lack this ability? We don’t know the answers to any of these questions.
For therapeutics, an important question I’m interested in is whether smaller ligands to the globular domain (say, a small molecule instead of an antibody) would be equally toxic. David Harris’ DS26 molecule was reported to bind to PrP’s C terminus but to actually enhance interactions with the N terminus, rather than blocking them [U.S. Patent Application WO2014025785A2]. And not all epitopes are toxic. The (non-linear) epitopes of the POM1 antibodies were originally reported in [Polymenidou 2008] and there is a nice tabular summary of what residues are bound and which epitopes are toxic in Table S2 of [Sonati 2013]. So I set out to plot in PyMOL which parts of PrP’s surface are toxic, benign, or unknown, according to that paper.
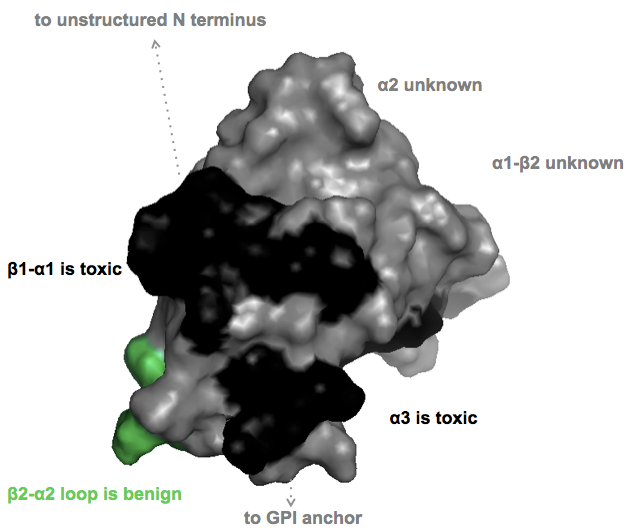
PyMOL code here
As you can see, for much of the protein’s surface, toxicity isn’t known. There are three reasons for this:
- The POM monoclonal series used in the toxicity studies doesn’t include antibodies to some of the epitopes
- The C-terminal POMs all have non-linear epitopes, so it’s not fully known which residues contribute to which epitopes
- Some of the toxic and non-toxic antibodies touch an overlapping set of residues. (Here, I’ve been conservative and have left the conflicted residues gray / “unknown”.)
From the perspective of wanting to design a small molecule to bind PrPC without being toxic, I think the short answer from the above diagram is it’s going to take a lot of empirics. While some epitopes are consistently toxic and one is consistently benign, most of the globular domain’s surface either hasn’t been characterized or is conflicted. This, together with the fact that we don’t yet know to what extent small molecules will mimic the toxicity observed with antibodies, to me says that we shouldn’t be shy about looking for PrPC binders, but we should be careful to characterize any that we find.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.