CJD2016

These are my plain-language notes from a few of the talks at the CJD Foundation’s 2016 family conference held in Washington, D.C. July 8-10, 2016. These notes are not exhaustive — I have omitted some talks that I missed or that are unpublished work the investigators did not want to make public yet.
Joel Watts

Dr. Watts provided an introduction to why and how we use mice to study prion disease. He noted that working with mice is a privilege, and that we’d all like to use fewer mice in research, but there are a few reasons why mice are still essential for studying prions:
- We don’t currently have a way to study human prion replication in cultured cells. There are some in vitro (cell-free) methods but they don’t recapitulate the whole story of what happens in the brain.
- The brain is a complex organ with multiple cell types which cannot yet be recapitulated in a dish. In particular, although some cultured mouse cells can replicate prions, these cells don’t die or seem to suffer any neurotoxicity due to being infected with prions.
- Some processes, such as prion neuroinvasion from the periphery into the brain, can only be studied in the whole animal.
- Drug development requires assessing a compound’s ability to cross the blood-brain barrier or avoid being metabolized by the liver. Living animals are the only models we have for this.
He then reviewed a few of the proofs of concept that we’ve learned from mouse models of prion disease. Mice genetically engineered to lack PrP (the protein that forms prions) cannot get prion disease [Bueler 1993]. Shutting off PrP production, even after mice are infected, can reverse the disease [Mallucci 2003]. An antibody against PrP can prevent peripheral (outside of the brain) prion disease from getting into the brain, although it doesn’t work once the disease is already in the brain [White 2003]. All of these findings tell us that PrP is the target we would like to be able to hit with a drug for prion disease. Note that this is very different from Alzheimer’s disease, for instance, where there are multiple genes and proteins involved in the disease, and it is not always clear which one is the right target for a drug. In prion disease, it is unequivocal that PrP is the protein essential for the disease, and if you can find a way to target it, you can treat the disease.
The usual mouse model of prion disease is mice infected with prions. People have developed several small molecules that can extend survival time in these mice, sometimes by double or more [Kawasaki 2007, Wagner 2013, Berry 2013]. One thing we’ve learned from these studies is that the earlier you treat prion disease, the bigger effect you can have — one compound, when given prophylactically before the prion infection, extended survival time by four-fold [Giles 2015]. Some of these molecules allow prions to evolve and develop drug resistance [Berry 2013], kind of like antibiotic-resistant bacteria, but some molecules don’t seem to cause drug resistance [Giles 2016]. Unfortunately, the reason none of these molecules are now in clinical trials is that although they work well against mouse prions, none of them work at all in mice genetically engineered to express human PrP and infected with human prions [Berry 2013, Lu & Giles 2013, Giles 2015, Giles 2016].
About 99% of studies of prions in mice use mice infected with prions, but infections only cause <1% of prion disease in humans. Most humans with prion disease develop it spontaneously, either by chance (sporadic prion disease) or due to a genetic mutation (genetic prion disease). Dr. Watts has therefore spent years working to create mouse models that develop spontaneous prion disease, as a closer analogy to prion disease in humans. The bank vole is a rodent that is uniquely susceptible to a wide variety of types of prions [Watts 2014], so Dr. Watts hypothesized that adding genetic mutations that cause disease in humans to bank vole PrP would result in mice that get spontaneous disease similar to that in humans. With support from CJD Foundation, he created mice that produce bank vole PrP with the D178N, E200K, or ΔGPI mutations, and characterized how these mice do indeed develop a spontaneous disease that is in many ways similar to genetic prion disease in humans [Watts 2016]. I have written a more detailed review of Dr. Watts’ new work here.
Zuzana Krejciova

Dr. Krejciova presented her work on developing a human cell model of prion replication — I have previously blogged about her work at Prion 2016. There is only one report in the literature of human prions propagating in cells [Ladogana 1995], but it wasn’t sustained over the long term. Because people studying prion replication in mouse cells have been able to discover compounds that successfully treat prion disease in mice, but don’t work against human prions [Berry 2013], she hopes that if we can develop a model where human cells propagate human prions, we could use them to find a compound effective against human prions.
She started by trying the “low hanging fruit” of a cancer cell line (glioma) that divides rapidly and produces lots of PrP, but she wasn’t able to infect them with prions. So she hypothesized that maybe non-dividing cells would be a more authentic model, since many/most cells in the adult human brain are non-dividing. She started with human induced pluripotent stem cells, differentiated them into astrocytes (a type of glia, a cell that helps neurons in the brain), and then made them stop dividing.
We refer to genetic changes in PrP by codon numbering — a codon is a position within the protein. I’ve written an introduction to the terminology here. There are two common versions of PrP in the general population - codon 129 can be M or V. Since everyone has two copies of the PrP gene, people can be MM, MV, or VV.
Dr. Krejciova found that she could get variant CJD (vCJD) prions to replicate in her astrocytes, as long as the cells had the same version of PrP. All cases of vCJD in humans have been in people who have MM at codon 129, and accordingly, she found that she could get the prions to replicate only in cells that were also MM.
This is an important step forward towards modeling human prions in cell culture. The procedure is currently fairly labor-intensive, the cells grow slowly, and can differ a bit from plate to plate of cells. So further work is required to get this model to a point where it could be used for discovering antiprion drugs.
Hermann Altmeppen

Dr. Altmeppen is based in Eppendorf, Germany, which is part of Hamburg. He studies the proteases — enzymes that cut up proteins, which he depicts as scissors or pac-mans — that cut PrP. There are two places that PrP normally gets cut — one is right in the middle of the protein, called alpha cleavage, and the other is at the base of the protein, called shedding. It’s called “shedding” because PrP is normally attached to the cell surface, and when it is cut it gets “shed” off of the cell.
Dr. Altmeppen studies an enzyme called ADAM10 that appears to be responsible for most or all of the shedding of PrP. In cells, if you delete the gene for ADAM10, you don’t get any PrP shedding. He wanted to study this in a mouse, but mice can’t live without ADAM10, so he used a specially engineered mouse that has ADAM10 deleted only in some cells at some times of its life. In these mice, there is little or no shedding of PrP, and they get prion disease more rapidly than normal mice [Altmeppen 2011, Altmeppen 2015]. This suggests that shedding is at least partially protective, and that maybe increasing shedding could be helpful. He is now investigating pharmacological approaches to encouraging shedding.
In Q&A, Dr. Altmeppen acknowledged that there is some risk in targeting PrP shedding, becuase genetically engineered mice that produce “anchorless” PrP that is never attached to the cell (kind of like if it was always automatically shed) can be infected with prions [Chesebro 2010, Stohr 2011]. He noted there is data suggesting that alpha cleavage is more protective [Westergard 2011], and might be the better therapeutic target. However, he said, the limitation is that we don’t have as much clarity on what enzyme(s) are responsible for alpha cleavage — some people say it is ADAM10 and/or ADAM17, but his view is that the jury is still out on this.
Byron Caughey
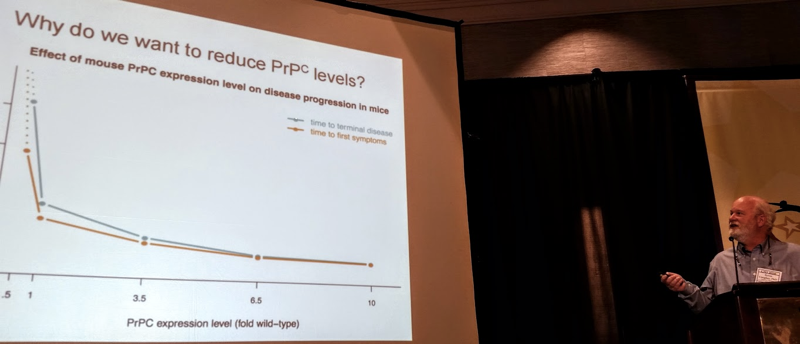
Dr. Caughey introduced the therapeutic strategy of reducing PrP levels to treat prion disease. He introduced several proofs of concept for this, which I have also discussed here and some of which Joel Watts mentioned earlier today. PrP is the protein that forms prions and it is required for prion disease. Mice that produce more than the normal amount of PrP develop prion disease very rapidly [Fischer 1996], while mice that produce half the normal amount of PrP can survive prion infection much longer than normal mice [Bueler 1994], and mice that produce no PrP can never get prion disease [Bueler 1993]. Meanwhile, it appears that reducing the amount of PrP in the brain is probably safe. Mice genetically engineered to produce no PrP have only mild health issues [Bueler 1992, Bremer 2010], and mice that produce half the normal amount of PrP are not known to have any health issues at all [Bremer 2010]. The few humans who have been identified to have only 1 working copy of the PrP gene, instead of the normal 2 copies, are healthy [Minikel 2016]. Based on all of these findings, reducing PrP levels seems to be a promising therapeutic strategy, and Dr. Caughey’s lab is working on pursuing this strategy.
Roberto Chiesa

Dr. Chiesa began by discussing the pros and cons of different therapeutic strategies for prion disease. Prion disease occurs when a protein called PrP changes shape: the normal protein, PrPC, converts to a misfolded form called PrPSc. Therapeutics to target PrPSc might work, but because PrPSc exists in a variety of conformations (shapes or ways of being folded), these also have the risk of being specific to only one strain or subtype of PrPSc, as Joel Watts noted. Dr. Chiesa therefore favors the strategy of targeting PrPC, to reduce it (as Dr. Caughey discussed) or stabilize it to prevent misfolding.
Dr. Chiesa investigated a series of porphyrin molecules and found one, called Zn(II)-BnPyP, that inhibits the formation of prions in protein misfolding cyclic amplification (PMCA), a procedure which uses sonic energy to amplify prions in vitro, in a cell-free system. He then tested it in prion-infected mouse cells, and found that it reduced the amount of misfolded PrP produced in these cells. To measure misfolded PrP, he used the enzyme proteinase K (PK), which can digest and break down normal proteins (including PrPC) but can’t break down PrPSc. In the process of doing this experiment, he also noticed that not only was PrPSc reduced in the cells treated with Zn(II)-BnPyP, but also, the amount of PrPC was reduced as well. He then did time-series experiments in mouse hippocampal neurons, and found that treating the cells with Zn(II)-BnPyP at concentrations of 5 or 10 micromolar over 24 hours almost completely removed PrP from the cells. The effect was reversible: if you remove the compound, the PrP comes back.
They then looked into how the compound works. They found that it doesn’t affect the levels of PrP RNA, so it doesn’t seem to affect how much PrP is produced. But when they treat the cells with chloroquine, a compound that blocks protein degradation, this prevents Zn(II)-BnPyP from having any effect on PrP levels. Therefore, they think Zn(II)-BnPyP reducesPrP levels by causing PrP to be degraded.
To test the compound in an animal model, they treated mice with 10 mg/kg (about 0.3 milligrams per mouse) of compound via intraperitoneal delivery (the compound was injected directly into the body cavity) three times a week for two weeks, and then measured both compound levels and PrP levels in the brains of the mice. Because the compound is fluorescent, they could assess whether it was in the brain by in vivo fluorescence imaging, as well as by a technology called HPLC. They found that the compound did seem to get into the brain, and there were about 2.9 micrograms of the compound per gram of brain tissue. Meanwhile, the PrP levels were reduced by about half. They looked for adverse side effects of the compound, but didn’t find any problems in the mice. They therefore think this could be a therapeutic candidate molecule worthy of further study, and they are currently planning experiments to test its effects on the survival time of prion-infected mice.
“Making a Difference” panel

Photo by Zuzana Krejciova.
Amanda Kalinsky, Trevor Baierl, Sonia, and I spoke about the ways in which we’ve reacted to our risk for genetic prion disease. Trevor made a documentary about his mother’s illness and his genetic testing journey, you can learn more on Facebook. Amanda has become an advocate for IVF/PGD and you can read more about her story in the New York Times. Sonia and I became scientists to work on developing drugs for prion disease, you can read more in the Boston Globe.
Ryan Maddox

Dr. Maddox provided an overview and update on the activities of the U.S. Centers for Disease Control and Prevention (CDC) on surveilling prion disease. Most of his talk was similar to his remarks last year, so I won’t go into all of the details here, but I wanted to highlight one important point he made. The incidence of prion disease is often quoted as 1 death or 1 new case per million population per year, though it may be higher, in the range of 1.5 or 2 per million per year. But because these figures are per year rather than lifetime risk per person, they make prion disease sound rarer than it is. The current estimate is that about 1 in every 6,000 deaths in the U.S. is caused by prion disease, meaning that for about 1 in every 6,000 people in the general population, prion disease will be their eventual cause of death.
Jiri Safar

Dr. Safar provided an overview of the activities of the surveillance center in Cleveland, and the improvements and upgrades they have made over the past few years. The most exciting improvement is the great increases in diagnostic accuracy brought about by RT-QuIC. A 5-year autopsy assessment of cases referred to the surveillance center revealed that end-stage clinical diagnosis of probable prion disease is only correct 61% of the time. Obviously, prion experts such as Brian Appleby and Michael Geschwind are the exception, as they are highly skilled in recognizing these diseases; the 61% figure reflects the relatively poorer understanding of prion disease by the average neurologist, who may only see 1 or 2 cases of prion disease in their career. It is therefore very important that the NPDPSC has dramatically improved its diagnostic capability in the past few years. Historically, cerebrospinal fluid protein markers such as 14-3-3 and total tau were used for diagnosis. The NPDPSC has now implemented second-generation RT-QuIC protocol [Orru 2015] which is sensitive enough to detect 21 attograms (an attogram is 10-18 grams) of PrPSc. Analysis of the patients tested so far by the surveillance center indicates that the test has >98% specificity and >92% sensitivity. This new level of diagnostic accuracy will be important for enabling clinical trials and the development of therapeutics.
In Q&A, I asked whether all referred cases are tested by RT-QuIC, or only cases that are considered positive by the older, less accurate tests of 14-3-3 and/or tau. Dr. Safar said that when they first implemented RT-QuIC a couple of years ago, only cases with at least slightly elevated tau levels were tested, because implementing RT-QuIC at scale poses a significant logistical and financial challenge. However, they are now ramping up their capacity for RT-QuIC, and they expect to soon be testing 100% of referred cases.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.