Why do some PrP mutations cause disease?
introduction
When I’ve given talks on prion disease genetics over the past year, one question I very often get is “why do these mutations cause disease?”. In other words, of all possible genetic variants in PRNP, what makes some benign, while others almost guarantee that a person will develop prion disease?
The short answer is we don’t know. The long answer is this blog post.
There are three functional classes of pathogenic variants in PRNP: protein-truncating variants, octapeptide repeat insertions, and missense variants. Let’s go through each class.
protein-truncating variants
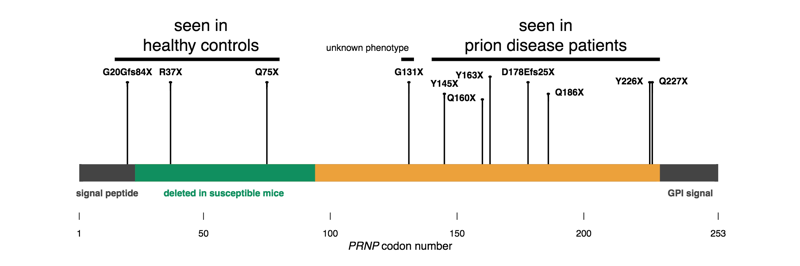
Figure 4 from [Minikel 2016]. PrP is encoded by a single exon; early truncating variants appear to be tolerated in humans, while late truncating variants cause prion disease.
I’m starting here because these are the simplest to explain — sort of. PrP is normally GPI-anchored to the outside surface of a cell. Protein-truncating variants occurring after codon 145 or so seem to leave enough of the protein intact that it is still capable of forming a prion, while lopping off the GPI anchoring signal, thus making PrP secreted instead of cell surface anchored. This change in localization causes a gain of function. Consistent with this, expression of full-length, GPI anchorless PrP in mice can cause prion disease too [Stohr 2011]. Truncation earlier in the protein doesn’t seem to cause any harm. Why secreted PrP is inherently bad is not obvious, not to me anyway. If anything, I might have imagined that letting PrP float around in three dimensions, instead of crowded into the 2D surface of a lipid raft, would give it fewer opportunities to bump into another copy of itself and form a prion. But while there are still some mysteries, we can at least point to a functional change in PrP caused by these mutations.
octapeptide repeat insertions

Cartoon depiction of the N-terminal toxicity theory.
These mutations increase the number of octarepeat units (PHGGGWGQ), which are involved in metal binding, among who knows what other functions. So these mutations increase the length of the flexible N terminus. A two-octapeptide repeat deletion has also been reported [Beck 2001, Capellari 2002], but there’s not yet strong human genetic evidence that it’s actually disease-causing. In fact, only insertions of at least 5 additional repeats appear to confer a high lifetime risk of prion disease — Mendelian segregation hasn’t been seen for shorter repeat insertions.
So far what we know is that adding at least 5 extra PHGGGWGQ units seems to be disease-causing. Why? The one explanation I’ve heard offered is what I call the N-terminal toxicity hypothesis, which holds that PrP’s N terminus has an intrinsic toxicity which under normal, healthy conditions is kept in check by alpha cleavage or by regulation by the C terminus, but which in disease can go unchecked. I haven’t heard a better theory, but at the same time I’m not totally satisfied by this theory either. For instance, the N-terminal toxicity hypothesis purports to explain an intrinsic toxicity of octarepeat expansions, independent of prion formation, yet these expansions also give rise to transmissible prions [Goldfarb 1991, Mead 2006]. In any event, even if the N-terminal toxicity hypothesis is correct, there’s still the question of how exactly the N terminus is toxic. For that, there are many ideas [reviewed in Aguzzi & Falsig 2012] but not much hard evidence yet.
missense variants
Close-up of the mutant asparagine in D178N-129M human PrP from [Lee 2010].
Now we come to the most perplexing part: why do PrP missense mutations cause disease? There has been a huge amount of speculation about this over the years and some interesting theories but no firm answers.
The most obvious question would be, do these mutations destabilize PrP’s native structure? If they do, they could increase the availability of a partially unfolded intermediate, thus providing fuel to PrPSc’s fire. But the best in vitro data on recombinant PrP with mutations provide no evidence for a categorical destabilizing effect. For instance, P102L and E200K HuPrP90-231 seem to have secondary structures, equilibrium guanidine denaturation curves, and melting points virtually identical to their wild-type counterpart [Swietnicki 1998]. A study of eight different reportedly pathogenic variants found that all of them had similar circular dichroism spectra as wild-type, and some but not all unfolded more readily in equilibrium urea denaturation [Liemann & Glockshuber 1999]. That study did include V180I and V210I, which we now recognize as lower-penetrance variants [Minikel 2016], and indeed, those two were among the variants with denaturation curves most similar to wild-type. But so was E200K, which clearly has pretty high penetrance. So our updated knowledge on this front doesn’t necessarily change interpretation of the data. A deeper study of F198S (a variant that does show Mendelian segregation with disease) found a clearly reduced stability in equilibrium denaturation experiments across a range of pH values [Vanik & Surewicz 2002], but without data indicating this is true across a range of different variants, it’s hard to say if this is the reason why F198S is pathogenic, or just a coincidence. There are even high resolution crystal structures of a couple of mutants, and while D178N and E200K PrP are apparently less likely to crystallize as a domain-swapped dimer than wild-type PrP, they otherwise have identical structures [Lee 2010]. So while a destabilization of native structure could be at play, so far, the in vitro experiments don’t provide any home-run evidence for it.
Recombinant PrP — unanchored, unglycosylated, and so on — could certainly fail to capture some folding problem encountered in the cell. There are reports to suggest that PrP with the D178N mutation, for example, is slightly unstable: its unglycosylated form doesn’t reach the cell surface and the protein is slightly under-expressed in human and knock-in mouse brains [Petersen 1996, Jackson 2009]. When bank vole PrP, which is already somehow unstable, has this mutation added to it, it takes tens of transgene copies to achieve even 0.5X expression in a mouse brain [Watts 2016]. But we don’t know how cells know to degrade these mutant PrPs (much less how we could encourage them to be even more vigorous about it).
Another possibility would be if mutations stabilized or promoted a misfolded structure, a hard thing to study directly since it is difficult to purify large amounts of authentically infectious PrPSc. A few studies have examined the propensity of recombinant PrPs to misfold, and have reported, for instance, that F198S is quicker than wild-type to form beta sheet-rich oligomeric structures [Vanik & Surewicz 2002], and that D178N is quicker to form RT-QuIC product [Gao 2012]. While interesting, there are a few difficulties in interpreting these sorts of studies. PrP amyloids formed in vitro are not usually infectious. Recombinant PrP properties can vary a bit from batch to batch even without sequence differences. As touched on above, it is hard to learn much from studies of single mutations because any one mutation might have unique properties by coincidence. By analogy, consider the case of SOD1 ALS, where as explained here, a subset of disease-causing mutants happen to cause a loss of function, even though the disease mechanism is clearly gain of function [Borchelt 1994, Hayward 2002].
A related and interesting theory (mentioned in the comments below) is that PrP mutations might stabilize some partially folded intermediate state, which is then on pathway to misfold. There is some evidence that PrP does encounter an intermediate state on its folding pathway [Apetri & Surewicz 2002] and that this intermediate state is more stable or more highly populated in certain mutant PrPs [Apetri 2004, Apetri 2006, Hart 2009]. But this could still be coincidental, as the presence of a folding intermediate is not perfectly correlated with a variant’s risk profile. For instance, in [Apetri 2004], the intermediate was highly populated for some high-risk variants like F198S but also for R208H, a variant we now believe to be low-risk at worst. Meanwhile E200K and P102L, both of which are highly penetrant, were similar to wild-type.
Relating to that N-terminal toxicity hypothesis from earlier, there’s some interesting evidence to suggest that PrP’s positively charged N terminus may dock on the face of the C terminus, and that D178N and E200K mutations, by increasing the charge on that face, may disrupt that interaction [Spevacek 2013]. If true, though, this still doesn’t do much to help us explain the mutations that don’t increase charge or aren’t located in that area.
One possibility is that there are a couple of different mechanisms at play — after all, genetic prion disease is fairly phenotypically diverse. For instance, several variants including P102L, G114V, A117V, and G131V are associated with a longer disease course (a few years as opposed <1 year for E200K). One theory about variants like those, which occur in the pretty hydrophobic central region of PrP, is that they might cause PrP’s cryptic transmembrane domain [Hay 1987, Lopez 1990, Harris 2003] to actually adopt a transmembrane conformation. The Prusiner and Lingappa labs had a series of papers about this so-called “CtmPrP” concept in the late 1990s [Hegde 1998, Hegde 1999] but interest in the topic eventually fizzled out without a definitive answer one way or the other.
There are other one-off explanations for certain variants as well, but they don’t get us very far. For example, T183A [Nitrini 1997, Grasbon-Frodl 2004] abolishes a consensus N-linked glycosylation site (residues 181-183 are NIT in the wild-type sequence), resulting in loss of one of PrP’s two potential glycan chains. But why, in turn, is losing one glycosylation site so bad, given that neither glycosylaton nor lack thereof seem to be critical for prion formation? The argument might be stronger if we’d ever observed a pathogenic variant abolishing PrP’s other glycosylaton site, residues 197-199. (F198S is located there but does not disrupt the consensus sequence NXT). And even if there were another example of a glycosylation site-abolishing variant, it seemingly would not get us closer to understanding how the many other missense variants, those that don’t affect glycosylation, cause disease.
There have been attempts to infer a mechanism of pathogenicity just from looking at the location and distribution of amino acid substitutions. For example, based on a predicted structure of PrPC (this was before the structure had been solved by NMR [Riek 1996]), one paper argued that pathogenic mutations must destabilize PrPC because “Ten of 11 known point mutations… lie within or adjacent to the four putative α-helical domains” [Huang 1994]. In a more recent example, it was argued that almost all pathogenic PrP variants either increase hydrophobicity or increase charge [Shen & Ji 2011].
There are a few issues to think about when evaluating such claims. What’s the null hypothesis that one is comparing to? For instanece, no two amino acids have exactly the same hydrophobicity, so one can guess that in any random, unselected distribution of amino acid substitutions, about half will increase hydrophobicity. (To model this formally, we’d have to also incorporate mutation rates based on DNA sequence context). So if half of pathogenic PrP variants increase hydrophobicity, that’s not surprising at all. And then, how many hypotheses is one testing? Amino acids have a number of properties (hydrophobicity, charge, size) and these can go up or down, so you have several degrees of freedom in trying to assign amino acid substitutions to different explanatory buckets. The same goes for location-based arguments. Is our prior assumption that missense substitutions in alpha helices should be the most destabilizing, or that those in loops and turns are the worst? Or maybe the key difference is interior vs. solvent-exposed residues? My point is that there are a lot of distributions of mutations that might occur by chance, but about which you could tell some post-hoc story that would sound pretty good.
Another limitation in these sorts of inferences is that we now recognize that not all of the mutations that were included in these analyses are actually disease-causing. The inclusion of some benign or low-penetrance variants could water down any conclusions quite a bit. Meanwhile, back then there weren’t a large number of known or presumed benign variants in PRNP to use as negative controls. For example, I’ve noticed that C214Y has turned up in gnomAD but still not in any prion disease case. I’d imagine abolishing PrP’s disulfide bond would be as destabilizing as anything, so this observation may slightly argue against the “destabilizing” hypothesis.
Per my effort to annotate the literature, there are only 16 PRNP missense variants with evidence for Mendelian segregation in at least one family: P102L, P105L, P105T, G114V, A117V, S132I, D178N, T183A, H187R, E196K, F198S, E200K, D202G, E211D, and Y218N. 11 of these variants lie in the structured region of PrP and, although I don’t think it will settle the matter of how or why these are pathogenic, I want to offer this map of where they lie in the protein:

To make this in PyMOL: fetch 1hjm; bg_color white; hide everything; show cartoon; color gray; color red, resi 132+178+183+187+196+198+200+202+211+218
Spinning this around in PyMOL, there’s no rhyme or reason to me. Helix 1 is spared, but other than that, you have interior residues and surface residues; helical residues and loop/turn residues. Some of the mutations increase charge, others are charge-neutral.
One could imagine much more sophisticated analyses and wet lab experiments here. But as I’ve dug into this more while writing this post, I think I’ve convinced myself that knowing the answer to this question is not critical, and depending on what the answer is, it might not move us any closer to a therapeutic. For example, suppose we made tens of recombinant PrP batches with various pathogenic mutations and various benign variants and did a huge battery of in vitro experiments on them, and suppose the result was we convinced ourselves that the “destabilizing” hypothesis is correct. Would that change our therapeutic strategy? You could argue that it would strengthen the case for finding small molecule PrP binders because it proves that lack of stability is the cause of disease. But you could also argue it weakens that case for finding small molecule PrP binders, because the mutant PrP molecules might already be unfolded and so the molecule won’t be able to bind them in the first place. On the whole it seems that time would be better spent working on actually finding small molecule PrP binders, so that you can test the therapeutic hypothesis with a candidate therapeutic agent.
In conclusion, we know that PrP mutations cause disease by a gain of function. We know that to cause disease, PrP needs to exist and it needs to misfold. These simple, longstanding observations are enough to lead us to some good therapeutic hypotheses. Why exactly some mutations cause disease and others don’t, we don’t know. And while I certainly care enough to read interesting work that others have done on the topic, I’ve realized I don’t think knowing the answer is critical to developing a drug.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.