A mechanism of action hypothesis

Crushed cinchona bark is the source of the natural product quinine, an antiprion compound whose mechanism of action is discussed in this post.
introduction
In the past couple of years, I’ve blogged a few times about proposed mechanisms of action (MoA) of known antiprion compounds. After some more pondering and several conversations with very smart people, I have a hypothesis about the MoA of a few of the compounds. From reading the literature, I can’t for the life of me tell whether this hypothesis is A) crazy, B) so obvious that everyone else already assumed it was true and hadn’t bothered to say it out loud, or C) somewhere in between. But in the hopes of finding out, I’ll share my thinking here.
background on antiprion compounds discussed in this post
This post is about quinine, quinacrine and their analogs and chlorpromazine. I will also touch on a few other antiprion compounds along the way, including protease inhibitors such as E-64d. All of these compounds have antiprion activity in cell culture, by which I mean, they reduce the amount of immunodetectable protease-resistant PrP produced in continuously dividing murine cell lines infected with mouse RML prions [Doh-Ura 2000, Korth 2001]. None of them seem to be active in vivo [Collins 2002, Barret 2003, Ghaemmaghami & Ahn 2009]. To the extent that it’s been studied, some of these compounds, such as quinacrine, don’t work in non-dividing cultured cells either [Ghaemmaghami & Ahn 2009]. Quinacrine also has strain-specific effects. For instance, it seems to actually promote CWD prion propagation, or at least protease resistance [Bian 2014], and it engenders drug resistance even among RML prions [Ghaemmaghami & Ahn 2009]. We don’t know whether any of these compounds are active against human prions because we lack a human prion-infected cell model and, as far as I know, no one has tested them in mice infected with human prions.
Given that these compounds don’t work in vivo, I’ve debated whether their MoA is interesting or important enough to spend time thinking about, but I’ve tended to come down on the side that this line of inquiry could yield some insights into prion biology and potential points for therapeutic intervention.
So what do and don’t know about the MoA of these compounds?
quinolines
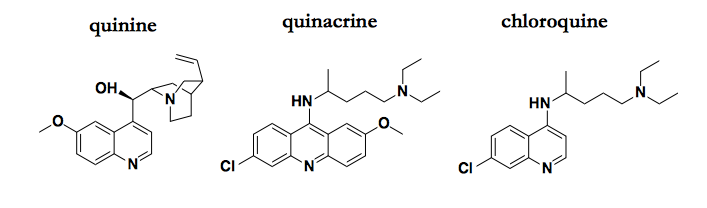
Just a few of the quinoline compounds that possess antiprion activity.
The antimalarial quinolines — quinacrine and its analogs, including the original natural product quinine — are probably the best-studied antiprion compounds, and several potential MoAs have been proposed, including binding PrPC, binding PrPSc, and redistributing cholesterol. All of these proposed explanations are unsatisfying, with a combination of weak evidence and low prior probabilities.
A key conversation that got me thinking about writing this post was when Stuart Schreiber pointed out to me that the cinchona tree spent millions of years developing quinine as a highly optimized defense against something, and that something was certainly not PrP. In general, natural products are exceptionally good at doing the one thing they evolved to do. For them to do a second thing equally well just by chance would be a fairly unlikely coincidence. So if they do a second thing well, Occam’s razor says the MoA for the second thing is probably somehow related to the first thing.
So what is that “first thing” that quinine evolved to do? And, by extension, that all its synthetic analogues, such as quinacrine and chloroquine, also do?
They alkalinize acidic compartments in cells.
These compounds all have the unique property of lysosomotropism. They are membrane-permeable as bases, but not as their conjugate acids. So they just drift around between different cellular compartments until they become protonated, at which point they’re stuck. As a result, they accumulate in the most acidic cellular compartment, soaking up protons, increasing the pH. It was shown decades ago that this property is required for the antimalarial effects of chloroquine, which accumulates in the P. falciparum’s acidic vacuole [Yayon 1984, Yayon 1985]. The parasite lives on human hemoglobin, but cannot digest heme, and must instead crystallize it into a structure called “hemozoin” to triage its toxicity [Slater & Cerami 1992]. Hemozoin forms spontaneously in acidic conditions but not in neutral conditions [Rush 2009]. So you could imagine how a base that gets into the vacuole and raises the pH would unleash the toxicity of heme, killing the parasite. Although, curiously, in the case of malaria, experts who know more than I do seem to think that the MoA is more complicated than that, and that alkalinization of the vacuole is necessary but not sufficient for efficacy of these compounds [Francis 1997, Foley & Tilley 1998]. Yet it really does appear that these compounds have no protein target, as the only known resistance mechanism in P. falciparum is the upregulation of xenobiotic transporters that pump the compounds back out of the vacuole [Fidock 2000]. If there were a protein target, one would expect the gene encoding that protein to be found mutated in some resistant isolates.
In mammalian cells, where hemozoin is not a factor, the main effect of raising lysosomal pH seems to be to inactivate proteases. Lysosomal proteases, like other secretory pathway proteins, are co-translationally translocated across the ER membrane and mature in steps through the ER and Golgi. Their inability to function at the neutral pH of those compartments is what keeps them from chewing up the whole secreted proteome [Cooper 2000 Chapter 9 - Lysosomes]. Instead, they only turn on when the compartment they are in matures into a lysosome and proton pumps turn on and acidify it. Thus, quinine et al inactivate lysosomal proteases. Apparently this is widely known. Last month I attended a talk totally unrelated to prions, where on one slide the speaker briefly touched on the effects of inhibiting autophagy in a particular cancer cell line. The slide named chloroquine as the tool compound used for this purpose, without further explanation, as if everyone in the audience simply knew this was how things worked.
Once I realized how widely accepted this was, I wondered, is this in fact precisely why people tested these compounds against prions in the first place? It appears the answer is probably yes.
The earliest reference I could find to the use of any quinine analog in the study of PrP or prions was in [Shyng 1993], the classic cell biology paper where David Harris first reported alpha cleavage and shedding of PrP. That study used pulse-chase, Western blots, immunofluorescence, and a few tool compounds to elucidate the cellular metabolism of PrPC in (uninfected) chicken cells. Pulse-chase showed that PrPC cycles repeatedly between the cell surface and an endocytic compartment, with about 5% of it undergoing alpha cleavage while in that endocytic compartment. When 300 μM chloroquine was applied to the cells, alpha cleavage was diminished. (As an aside, already in 1993, the fact that chloroquine alkalinizes acidic compartments was so well-known that it was mentioned without citation.) Ammonium chloride (NH4Cl) and leupeptin also inhibited this cleavage event. Ammonia is, like quinine and quinacrine, a lysosomotropic weak base that can alkalinize acidic cellular compartments, and leupeptin is an inhibitor of cysteine, serine, and threonine proteases — both are considered tool compounds for inhibiting lysosomal function [Seglen 1983]. Aprotinin, another protease inhibitor, also inhibited the cleavage, whereas EDTA (to chelate metals and thus inactivate metalloproteinases) and pepstatin (an aspartyl protease inhibitor) had no discernible effect, so the conclusion was that alpha cleavage was performed by pH-sensitive serine proteases in an acidic endocytic compartment. (I’m not sure how this squares with the nowadays conventional wisdom that alpha cleavage is performed by ADAMs, which are metalloproteinases [Liang & Kong 2012]). The study never touched on PrPSc, but it established that PrPC does normally cycle between the cell surface and someplace acidic inside the cell, and that this cycling is part of its endogenous metabolism.
The next time these compounds turn up is when Katsumi Doh-Ura and Byron Caughey test quinacrine and chloroquine in prion-infected ScN2a cells several years later [Doh-Ura 2000]. They don’t cite the Shyng paper, but they do refer to the compounds as “lysosomotropic agents”, and they cite work from the Caughey lab indicating that PrPSc undergoes beta cleavage in the lysosome [Caughey 1991] (more on this below). In addition to the lysosomotropic compounds, they also test the cysteine protease inhibitors leupeptin, E-64, and E-64d. So although the hypothesis / reason for interest in these compounds is never explicitly stated, the thought process seems to be similar to David Harris’ thought process: pH-sensitive proteases are doing something, and here are two orthogonal ways to inactivate them. Doh-Ura et al contemplated a few different hypotheses as to why the inactivation of these proteases might be inhibiting prion propagation. For instance, in one model, there exists a chaperone that breaks down prions, but that chaperone is itself degraded by lysosomal proteases, so inhibiting the proteases results in increased destruction of prions.
chlorpromazine
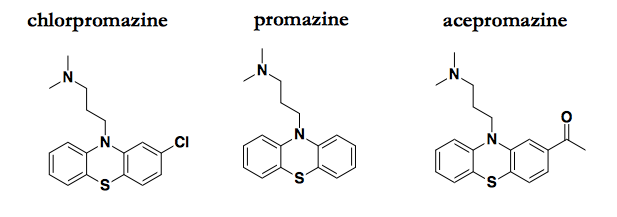
Chlorpromazine and a couple of analogs that also possess antiprion activity.
I have discussed the literature on these compounds in a few previous posts. Chlorpromazine’s antiprion MoA was largely unspeculated-upon until 2014, when a crystal structure was published showing chlorpromazine bound to PrPC [Baral 2014]. At the time, I took this at face value and entertained the possibility that PrPC binding might really mediate chlorpromazine’s antiprion effects. But having learned more about both chemical biology and structural biology over the past two years, I now consider that explanation very unlikely. While crystallography is a powerful tool for determining the exact binding mode of known ligands, it suffers from an excess of sensitivity: the conditions of crystallization are so extreme that compounds can be seen bound to a target for which they bear no affinity measurable by any other means. Meanwhile, chlorpromazine is quite potent against prion infection in cell culture, with low micromolar efficacy (EC50 ≈ 3 μM [Korth 2001]) whereas chemical shift changes in NMR were only observed at millimolar concentrations (6-12 mM promazine [Baral 2014]). And of course, the prior probability of chlorpromazine binding PrPC with affinity sufficient to explain its bioactivity seems very low considering that chlorpromazine was not invented with PrPC in mind.
Here too, a key conversation has shaped my thinking. This conversation was over breakfast with Emiliano Biasini one morning at Prion2015. He rejected out of hand the notion that chlorpromazine binds PrPC, and pointed out that cell biologists use chlorpromazine as an inhibitor of endocytosis, so that has to be the way it works for prions.
Chlorpromazine, mind you, wasn’t developed as an endocytosis inhibitor. It is the original “typical antipsychotic” drug developed to treat schizophrenia [Meltzer 2013], and most people seem to agree that its mechanistic target in the treatment of that disorder is the D2 dopamine receptor, encoded by DRD2 [Hyman 2012]. At first glance, the fact that a drug developed for the dopamine receptor also happens to inhibit endocytosis seems to contradict my Occam’s razor rule discussed for quinine above, but apparently it’s all a matter of potency.
One classic paper [Creese 1976] measured chlorpromazine’s potency against dopamine receptors indirectly through displacement of radiolabeled haloperidol, which is another, more potent, typical antipsychotic with reported a KD of 2 nM for dopamine receptors. That paper reported a Ki value of 10 nM for chlorpromazine. A more recent work I came across reported that chlorpromazine has a Ki of 4 nM for the D2 dopamine receptor [Kroeze 2003]. But chlorpromazine is highly promiscuous: that paper also showed that it has nanomolar affinities for a dozen other receptors, including serotonin and histamine receptors (see this table), some of which appear to underlie the drug’s notorious side effects [Kroeze 2003].
In contrast to all of those nanomolar values, inhibition of endocytosis only occurs when you get into the micromolar range. The original papers showing that trifluoperazine (an analog of chlorpromazine) interferes with receptor recycling [DiPaola 1984] and then that chlorpromazine blocks endocytosis of the LDL receptor [Wang 1993] used concentrations of 20 and 30 μM. The mechanistic target of chlorpromazine with respect to endocytosis is, therefore, not the D2 dopamine receptor, but presumably some other target for which it has three orders of magnitude weaker affinity.
That other target might be the dynamins, a group of proteins (encoded by DNM1, DNM2, and DNM3 in humans) that are required for scission (cutting off) of membranes during endocytosis [Sweitzer & Hinshaw 1998, Hinshaw 2002]. Emiliano Biasini sent me an interesting paper published last year showing that chlorpromazine and a few other atypical antipsychotics inhibit dynamin’s GTPase activity in vitro with EC50 values on the order of 10 μM [Daniel 2015]. The structure-activity relationship among the molecules is compelling: their potency at inhibiting dynamin GTPase activity is correlated with their potency at inhibiting endocytosis in cells, and both of those values are uncorrelated with their potency as dopamine antagonists [Daniel 2015].
So chlorpromazine, originally a drug for the dopamine receptor, happens to bind some other target, maybe dynamins, with much weaker affinity, and is thus a micromolar inhibitor of endocytosis. And “inhibitor” may not be quite the right word, because what the drug actually does is complicated. When fibroblasts were treated with chlorpromazine, the LDL receptor was visualized as being redistributed away from the cell surface and trapped in some intracellular compartment [Wang 1993]. So what chlorpromazine really seems to do is to introduce a blockade and a pileup at some particular step in the endocytic pathway.
In further support of this being the antiprion MoA of chlorpromazine, I emailed with Emiliano Biasini again while writing this post, and he gave me permission to share here that he now has a manuscript in preparation showing that chlorpromazine does indeed cause trapping of PrPC in intracellular compartments.
a review of some cell biology
What I think I know so far, then, is that chlorpromazine inhibits prion formation by introducing a blockade in the endocytic pathway, and quinacrine inhibits prion accumulation by inactivating acid-dependent proteases. How do we interpret this information? In other words, why would these MoAs affect prion propagation?
To try to get to the bottom of this, I started reading up on the cell biology of PrP, of endocytosis, and of lysosomes,. Cell biology is not my strong suit, so I’ve probably gotten some things wrong here, and please let me know if you find mistakes.
A few useful resources included an NCBI Book [Cooper 2000] and a couple of reviews [Maxfield & McGraw 2004, Grant & Donaldson 2009]. All of these contain diagrams of the endocytic pathway, minor variations on the same general picture. Here’s my very simple rendition of roughly what that diagram looks like:
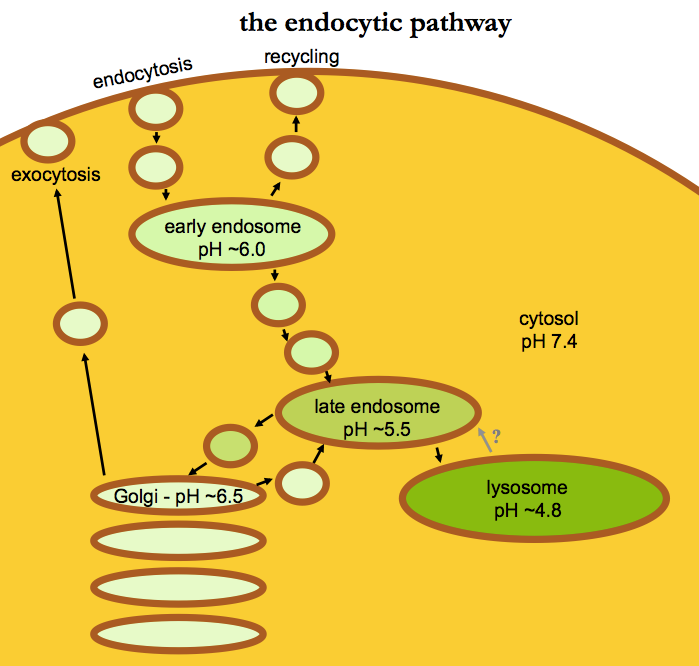
Disclaimer: I’m not a cell biologist.
As a general note before we move on, I just want to acknowledge this diagram is quite a simplification. The term “early endosome” includes at least two different organelles, a sorting endosome and an endocytic recycling compartment. I haven’t depicted multivesicular bodies. And as Maxfield & McGraw point out, even the organelles that are depicted here can be a moving target. What we call “late endosomes”, for instance, might be defined operationally by staining for a particular protein marker, but at the end of the day, they are “a heterogeneous collection of compartments with different properties, and the functional consequences of this heterogeneity are not well understood” [Maxfield & McGraw 2004].
A major role of the endocytic pathway is the degradation or recycling of cell surface receptors — in fact, a lot of this pathway was worked out through the study of the LDL receptor. First I’m going to walk through some key points from [Cooper 2000 Chapter 12 - Endocytosis] to make sure I understand them. When a ligand binds a receptor, it can undergo endocytosis, where the endocytic vesicles first fuse with early endosomes, which have an internal pH around 6.0-6.2. This pH is low enough to cause many ligands to dissociate from their receptors, such that the ligand can continue its journey inward into the cell while the receptor may be recycled back to the cell surface. This pH, however, is not low enough to lead to activation of any proteases. Some contents of the early endosomes can then be transported, in carrier vesicles, to the late endosomes deeper inside the cell. These have a lower pH of 5.5-6.0 and also accept infusions of transport vesicles directly from the Golgi, containing lysosomal acid hydrolases. “Acid hydrolases” simply means enzymes that break covalent bonds (hydrolases) and that are active only at low pH (~5.0). As the late endosomes further mature and acidify to pH ~5.0 with help of the V-ATPase proton pump, the hydrolases awaken, contents begin to be degraded, and people begin to call the organelle a lysosome. If you don’t like my diagram, Figure 12.41 is another useful depiction.
People always talk about recycling from the early endosomes back to the cell surface, but the textbook view of the endocytic pathway does not often include recycling from late endosomes or lysosomes back to the cell surface. For instance, in Figure 1 of [Grant & Donaldson 2009], the late endosomes are depicted as a dead end. Figure 9.36 of [Cooper 2000] depicts the cation-independent mannose-6-phosphate receptor (CI-MPR) — receptors for the signal that targets acid hydrolases from the Golgi to the late endosome — as recycling out of late endosomes, but only back to the Golgi, not the cell surface. Of course, the Golgi are also part of the secretory pathway, and not just the endocytic pathway, which is why in my diagram above I’ve drawn an arrow for exocytosis, the process by which secreted and membrane proteins get to the cell surface. If you were an enterprising protein in the late endosome looking to avoid a fate of being degraded in the lysosome, you can imagine a loophole where you hop a vesicle back to the Golgi, and then from there find a way to get exocytosed back to the cell surface.
Do any proteins actually exploit such a loophole? Figure 2 of [Maxfield & McGraw 2004] suggests this possibility and more, by showing a bidirectional arrow between late endosomes and lysosomes (which I’ve included in gray with a question mark above), and then a network of arrows that in principle lead from late endosomes to the trans-Golgi network, then to the endocytic recycling compartment, and finally to the cell surface. The text mentions two examples of proteins that can traverse this whole pathway. One protein, furin, recycles repeatedly between the late endosomes and the trans-Golgi; they state that furin’s “net rate of export back to the cell surface is low”, but “low” presumably means non-zero. The other protein is CI-MPR itself, and like furin, its “net rate of… return to the cell surface is low.” So according to this source, it is at least possible, though perhaps not common, for proteins from at least the late endosomes to recycle back to the cell surface. Curiously, that bidirectional arrow between lysosomes and late endosomes is never explained, so it is still not clear to me whether it is possible to get out of the lysosome and back to the cell surface.
A brief detour before we move on: at the beginning of this section I glossed over how endocytosis occurs. The reviews cited above focus mostly on clathrin-mediated endocytosis, where the protein clathrin forms a lattice that physically deforms the membrane, causing it to bubble inward. This is one area where PrP may or may not be special. It is generally held that GPI-anchored proteins are endocytosed by clathrin-independent mechanisms [Dutta & Donaldson 2012], which seems intuitive, since clathrin is cytosolic, and GPI-anchored proteins such as PrP lack a cytosolic domain by which to signal to it. Yet there are also several lines of evidence to indicate that PrPC does undergo clathrin-mediated endocytosis [Harris 2003]. There seems to be no consensus in the literature, and the introduction of one paper I read says “there is no consensus” [Kang 2009].
cell biology of prion propagation
Earlier in this post I touched on some of David Harris’s early work characterizing the normal cellular metabolism of PrPC [Shyng 1993]. A number of people in the early 1990s were also working on understanding the cell biology of when, where, and how PrPSc was formed, and several discoveries supported the notion that PrPSc was to be found somewhere along the endocytic pathway.
The Caughey and Prusiner labs had both discovered that PIPLC, an enzyme that can cleave GPI anchors, would release PrPC, but not proteinase K-resistant PrPSc, from the surface of prion-infected cells, thus implying that whereas PrPC could be found on the cell surface, PrPSc was buried somewhere inside the cell [Caughey 1990, Borchelt 1990, Taraboulos 1990]. Similarly, PrPC, but not PrPSc, could be labeled by sulfo-NHS-biotin, a cell-impermeable biotinylation reagent, again implying PrPSc was intracellular [Borchelt 1990]. It was also found that PrPSc co-localized at least partly with fluorescently labeled wheat germ agglutinin (background here), a lecithin that binds N-acetyl-glucosamine, considered a marker of the Golgi [Taraboulos 1990]. In contrast, there is also an obscure paper for which I can’t find full text [McKinley 1991] where they apparently did electron microscopy and found PrPSc localized in “secondary lysosomes” and not Golgi. (Secondary lysosomes must be a term that has fallen from favor, because there weren’t many recent Google hits for it; it seems to refer to lysosomes that have fused with phagosomes or other vesicles bringing in cargo from the cell surface).
The Caughey lab also characterized the N-terminal truncation (what we now call beta cleavage) of PrPSc in cultured cells, and found that this proteolytic event (just like alpha cleavage) could be inhibited by either ammonium chloride (NH4Cl) or leupeptin [Caughey 1991]. The fact that PrPSc was truncated, but not destroyed, by lysosomal proteases was interpreted to mean that PrPSc did encounter the lysosome, but only after its acquisition of protease resistance [Caughey 1991].
A caveat to all of these studies is that they only considered proteinase K-resistant PrPSc, as this was years before the existence of protease-sensitive prions was demonstrated [Safar 1998]. Nevertheless, these studies certainly showed that at least some PrPSc is found in an intracellular compartment somewhere along the endocytic pathway. This left the question of whether the substrate for conversion to PrPSc is is newly synthesized PrP just headed out to the plasma membrane for its first time ever (and getting intercepted by PrPSc in the Golgi, perhaps), or PrPC that has already reached the cell surface and then been re-internalized. Early experiments showed that pulse radiolabeling PrPC and then treating with PIPLC prevents the appearance of radiolabeled PrPSc, indicating that cell surface PrPC is incorporated into PrPSc [Caughey & Raymond 1991, Borchelt 1992]. Indeed, subsequent experiments showed that longer treatments with either PIPLC or monoclonal antibodies to PrPC can cure infected cell cultures [Peretz 2001, Enari 2001], suggesting that, at least in ScN2a cells, re-internalized PrPC is the predominant substrate for conversion to PrPSc, with little or no contribution from nascent PrP just coming up the secretory pathway.
As an aside, while reading all this literature, I noticed that some of the same treatments used to understand the metabolism and localization of PrPC or PrPSc also reduce PrPSc accumulation in cells. At first I was troubled as to how these results could be interpretable. For instance, how can you study differences in the N-terminal truncation of PrPSc upon leupeptin treatment if leupeptin also makes PrPSc go away altogether? The difference, it turns out, lies mostly in the length of time for which cells are exposed to the compounds. For instance, leupeptin’s impact on alpha cleavage was demonstrated after a 16 hour treatment [Shyng 1993] and its impact on beta cleavage was demonstrated after a 20 hour treatment [Caughey 1991], whereas its impact on PrPSc levels was only demonstrated after a 4 day treatment [Doh-Ura 2000]. Similarly, PIPLC releases PrPC but not PrPSc from the cell surface after 5 hours [Taraboulos 1990], but it does eventually cure cells of PrPSc infection (presumably by depletion of PrPC substrate) after 3 days [Enari 2001].
interpretation
With all of that background, what is our interpretation? How can we put these puzzle pieces together into a coherent story about what quinacrine and chlorpromazine are doing to reduce prion formation?
A fairly minimalist explanation would be that PrPSc formation occurs in an acidic endocytic compartment and its formation requires pH-dependent protease activity. But what exactly do we mean by “formation”? In my early days of trying to understand what prions are, I internalized this simplistic vision that autocatalytic templating meant the equation PrPC + PrPSc → 2 PrPSc. I later learned that this is referred to as the heterodimer model:
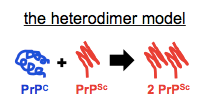
While I still use a graphic like this occasionally in slides when I am introducing the concept of autocatalytic post-translational conformational change to people who aren’t familiar with it, I think most prion scientists agree this isn’t really what goes on. If you can only ever add molecules of PrPSc, then how do you ever get exponential growth? We know that prion titers grow exponentially, so it must be that the fibrils also break to create more sticky ends for recruiting more monomers. Enter the breakable filament model (see for instance [Knowles 2009, Aguzzi & Falsig 2012]):
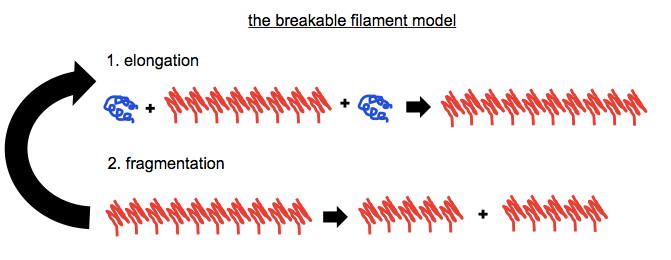
We know that this is how it works for the [PSI+] prion in yeast, with the chaperone Hsp104 responsible for doing the fragmenting [Chernoff 1995] — see this backgrounder. We also know that creating any appreciable titer of infectious PrPSc in vitro requires sonication (in PMCA) to do the fragmenting. Even to efficiently generate non-infectious PrP amyloid in vitro, for instance in RT-QuIC, requires shaking, perhaps to fill the same role. But no one seems to know exactly which proteins do the fragmenting of PrPSc fibrils in vivo.
With that framework in mind, do quinacrine and chlorpromazine affect elongation, fragmentation, or both? Many explanations are possible, but I reason as follows.
I find it relatively difficult to imagine why the activity of lysosomal proteases would be required for the elongation step, because A) what would they do? and B) because if active proteases are found in the same time and place as not-yet-converted PrPC, why don’t they just digest it? Instead, I find it relatively easy to imagine that proteases are involved in fragmenting PrPSc. Granted, PrPSc fibrils are held together by hydrogen bonds, not covalent bonds, so perhaps it would be more satisfying if some sort of “disaggregase”, rather than a protease, were responsible for the fragmentation. But couldn’t partial proteolysis, some sort of nicking of the largely protease-resistant fibril backbone, be enough to lead to fibril fragmentation? I speculate.
And if proteases are involved in fragmentation, then elongation must happen in a different compartment, because otherwise (as argued earlier), PrPC is protease sensitive and the proteases would just digest it. So I arrive at a model where elongation happens in one compartment, and then the elongated fibrils must be trafficked inward to an acidic compartment where pH-dependent proteases fragment them, and then the fragmented fibrils must re-emerge to undergo elongation again. We don’t know exactly which compartment is the site for either of these processes, and as noted above, definitions of the various organelles can sometimes be a moving target anyway. But for the sake of illustration, here are a couple of ways this could look.
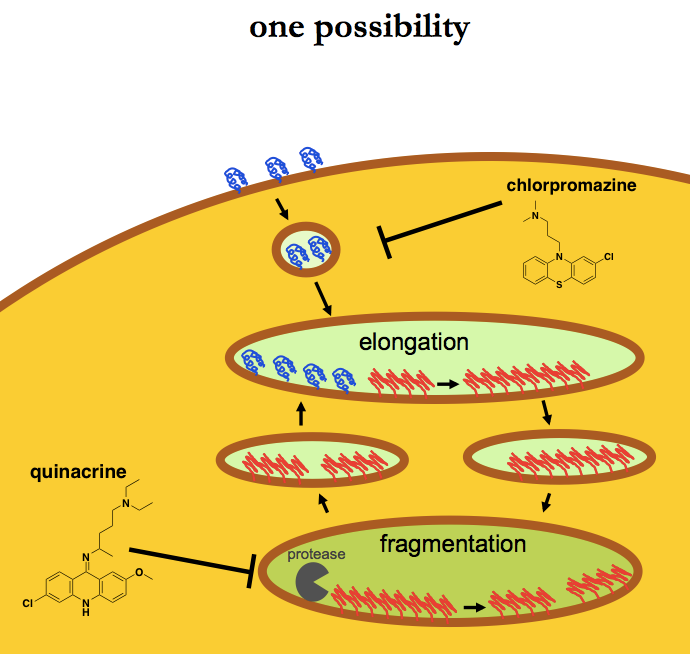
In this first model, endocytosis is required to deliver PrPC to an internal compartment where elongation takes place. Chlorpromazine works because it keeps PrPC from getting to where elongation occurs.
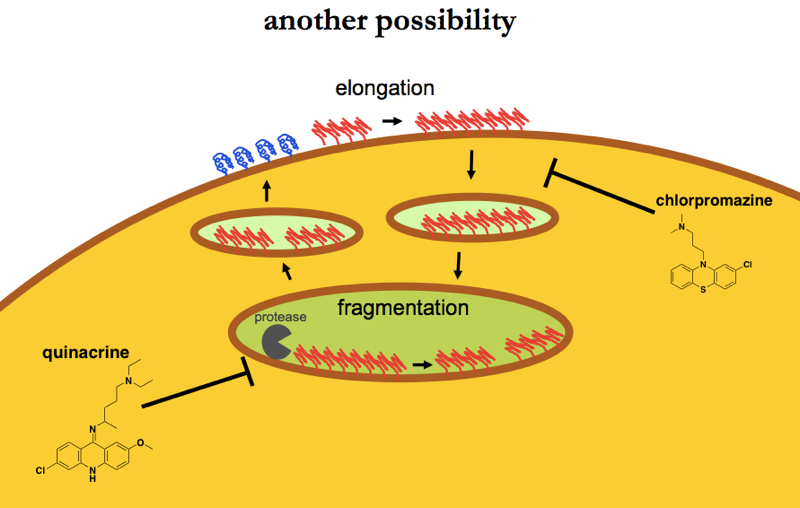
In this second model, elongation occurs on the cell surface, and endocytosis is required to internalize elongated fibrils for fragmentation in an internal compartment. Chlorpromazine works because it keeps fibrils from getting internalized and fragmented.
In either case, the exact identity of the acidic compartment where fragmentation hypothetically takes place is unclear. From all the reading I’ve done, I’ve only ever heard mention of acid hydrolases that become active at lysosomal pH, so pH ≤ 5.0 or so — see for instance [Cooper 2000 Chapter 9 - Lysosomes]. I have never heard of a protease that is pH-dependent yet which activates at a higher pH threshold such as 6.0 or 5.5. For this reason, I’m slightly inclined to think that the fragmentation occurs in lysosomes. On the other hand, as discussed above, it’s not clear whether proteins ever escape from the lysosome alive, so to suggest that prions fragment in the lysosome and then get back out to the cell surface might be heresy.
This general model, I believe, is consistent with the strange properties observed for quinacrine treatment: the fact that it is strain-specific and engenders drug resistance [Ghaemmaghami & Ahn 2009]. Different prion strains might have different degrees of “frangibility” (how easily they break). So perhaps upon chronic quinacrine treatment in non-dividing cells, a prion strain emerges that is more brittle and more readily breaks without the help of lysosomal proteases. That would make some sense in light of observations in yeast: Jonathan Weissman’s lab has shown that [PSI+] prions differ in how good they are at elongation and at fragmentation, and in their sensitivitiy to Hsp104 overexpression [Tanaka 2004, Tanaka 2006].
how to test it
This is all just a hypothesis. How could we determine if these are indeed the MoAs of chlorpromazine and quinacrine? I have done a bit of thinking on this, though I don’t necessarily have the most thorough answer yet.
One basic question I’d want to answer first, a step zero of sorts, would be to test whether these compounds actually reduce the number of propagons in cells (this hypothesis would imply yes), as opposed to just reducing the protease resistance of prions (a plausible alternative). As far as I know, all the reports of activity of these compounds have been based on proteinase K-resistant PrPSc as a readout; I don’t know that anyone has done bioassay, or scrapie cell assay, or RT-QuIC, or any other measure of seed number. Treating cells with these compounds and doing RT-QuIC on the cell lysates would be easy enough to do.
Then, my first idea would be to test a battery of tool compounds that have similar bioactivities, and see if they also inhibit prion accumulation. For instance, there are a whole series of trademarked small molecule dynamin inhibitors marketed to inhibit endocytosis (see for instance abcam’s collection or the list in the introduction of [Daniel 2015]), although the specificity of some of them has been called into question [Park 2013]. People also use a dominant negative mutant of dynamin 1 (K44A) to perturb endocytosis — in fact, it’s been used to study trafficking of PrPC [Magalhaes 2002]. Likewise, there are orthogonal ways to raise lysosomal pH, such as the V-ATPase inhibitors bafilomycin A1 or BRD1240 [Aldrich & Kuo 2015].
Note that NH4Cl has already been tested and found not to inhibit prion propagation in ScN2a cells, although this could just be for lack of potency [Doh-Ura 2000]. Which brings us to a bit of a problem with the experimental plan I have outlined so far — the experiments all have a bit of a confirmatory flavor to them, where it would be easy to get excited about the results that agree with the hypothesis while waving away those that don’t. Ideally I would like to also take a more negative approach and think of what would be the quickest experiment to disprove this hypothesis, but I haven’t come up with a good answer yet.
I’d also like to learn more details about which steps and compartments in the endocytic pathway are involved, but I’m not sure how far one could go with this. Certainly there are a battery of different protease inhibitors out there to see which proteases are involved. There are also inhibitors of closely related pathways, for instance, wortmannin, LY294002, and 3-methyladenine inhibit PI3K, blocking autophagosome formation [Wu & Tan 2010]. But from Googling around I haven’t found much in the way of probes for specific steps in the endocytic pathway in particular.
Is it worth developing these ideas further and doing some experiments? I’m not sure yet. As mentioned at top, neither quinacrine nor chlorpromazine actually works in vivo against any prion strain as far as we know, so I debate whether understanding their MoA is A) an irrelevant distraction, or B) critical for understanding why they don’t work and for finding compounds that do. The “B” side of me wrote this post, but the “A” side of me is wary of putting more time in. Maybe you, the reader, have suggestions for how to prove, disprove, or improve this hypothesis, and maybe your suggestions will make it worthwhile to take some next steps.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.