The curious antiprion activity of antimalarial quinolines
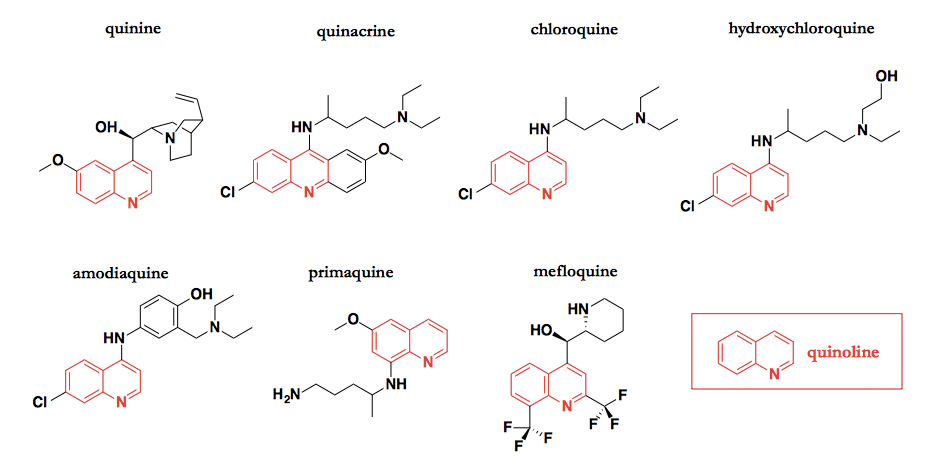
Quinoline derivatives are some of the oldest drugs around. They all stem ultimately from quinine (top left), a natural product isolated from the bark of the Andean cinchona tree, and indeed, according to Oxford Dictionaries the word quinine comes from the Quechua word kina, for bark. Quinine isolated from cinchona bark became the first antimalarial drug by the early 1800s [Institute of Medicine 2004], and indeed, remained the only antimalarial for over 100 years. In the last century, many synthetic analogues were shown effective, including quinacrine [Green 1932], chloroquine [Loeb 1946], and mefloquine [Trenholme 1975]. All of these analogues retain the heterocycle highlighted in red, which is known as quinoline, although people generally refer to quinacrine as being a 9-aminoacridine compound, for the larger ring system of which the quinoline is but a substituent. Although there now also exist structurally distinct antimalarial drugs such as artemether, some quinoline compounds including chloroquine (Aralen®) and mefloquine (Lariam®) are still recommended today.
Despite the long history of these compounds, my read of the literature - and I’m no malaria expert, so let me know if I’m wrong - is that we still have little concrete understanding of the mechanism of action of quinoline derivatives [Foley & Tilley 1998]. It seems like what we do know is that when malaria parasites are treated with chloroquine, their food vacuoles expand and become filled with high concentrations of chloroquine [Aikawa 1972]. Various theories hold that chloroquine then interferes with lysosomal degradation of the parasite’s food supply by inhibiting proteases or phospholipases, or that it inhibits the parasite’s ability to sequester the heme molecules from digested hemoglobin into polymers called hemozoin to trap their harmful free radical activity [Foley & Tilley 1998]. The nice thing about pathogens with nucleic acid genomes is that now you can sequence them to determine the genetic basis of drug resistance. Frustratingly, however, quinoline drug resistance has been attributed not to mutations in some target, but rather, to mutations in genes encoding transmembrane transporters that appear to reduce the accumulation of the drugs in food vacuoles [Sidhu 2002, Price 2004].
Even as the mystery of the quinolines’ antimalarial mechanism has remained unsolved, a new set of mysteries has emerged concerning their anti-prion activity. Quinacrine is by far the most famous, and even went to clinical trials for prion disease throughout the 2000s. But of the 15 FDA-approved antimalarial drugs, there are 7 (shown above) that contain quinoline, and all 7 of these are reported to have a micromolar or lower EC50 for clearing proteinase K-resistant PrPSc from ScN2a cells:
| compound | antiprion EC50 | references |
|---|---|---|
| quinine | 6 μM | Murakami-Kubo 2004 |
| hydroxychloroquine | 1-10 μM | Kocisko 2003 |
| chloroquine | 2.3-10 μM | Doh-Ura 2000, Korth 2001, Kocisko 2003, Klingenstein 2006b |
| amodiaquine | 500 nM | Kocisko 2003 |
| quinacrine | 300-400 nM | Doh-Ura 2000, Korth 2001 |
| mefloquine | 500 nM | Kocisko & Caughey 2006 |
| primaquine | <10 μM | Kocisko & Caughey 2006 |
Quinacrine’s cellular EC50 of ~300 nM is among the lowest ever reported for an antiprion small molecule, and this catapulted it onto the world stage as a candidate drug for prion disease in the early 2000s. Its roundly disappointing history is narrated in this post, which I wrote almost three years ago. Briefly: the story begins with Katsumi Doh-Ura and Byron Caughey testing a panel of “lysosomotropic agents” and finding that several of them inhibit the accumulation of proteinase K-resistant PrPSc in cultured mouse neuroblastoma cells infected with RML prions [Doh-Ura 2000]. The most potent among these compounds is quinacrine, with an EC50 of just 400 nM. The results are replicated shortly thereafter by Carsten Korth in the Prusiner lab at UCSF, who reports an EC50 of 300 nM for quinacrine, and finds that several other antimalarial quinolines also possess antiprion activity, albeit with higher EC50s: ~4 μM for pamaquine and chloroquine. While a few other compounds with antiprion activity in cell culture or in vivo were already known at that time, and a few were even approved drugs, all of them were either quite toxic (amphotericin B), or were large, heterogeneous, hydrophilic polymers couldn’t cross the blood-brain barrier (pentosan polysulfate). To be sure, quinacrine has its own share of adverse events, and in fact it’s not even marketed in the U.S. anymore. But when pitted against a rapidly fatal untreatable disease, its toxicity was an easy tradeoff to accept, and accordingly, investigators at UCSF and elsewhere immediately launched into compassionate use trials in patients with sporadic prion disease. The first raft of studies indicated that quinacrine didn’t seem to be helpful in compassionate use trials in humans [Haik 2004], and that the drug had no survival benefit in animal models either [Collins 2002, Doh-ura 2004]. This disappointing outcome was eventually confirmed in clinical trials in the U.K. and U.S. [Collinge 2009, Geschwind 2013].
While it has been years since quinacrine seemed promising as an antiprion drug candidate, I periodically find myself wondering whether there is something fundamental about prion biology and therapeutics that we could come to understand by knowing what quinacrine’s mechanism is in cell culture and why it doesn’t work in vivo.
As a quick review, it is worth understanding the assay that has stood at the center of antiprion drug discovery efforts for over a decade. In this assay, mouse neuroblastoma cells infected with mouse RML prions (dubbed ScN2a cells, brief history here) are incubated with test compounds for 3-6 days, lysed, digested with proteinase K, and then immunodetection (which might mean Western blot, dot blot, or ELISA) is used to quantify the remaining, and therefore protease-resistant, PrP. This procedure was the basis of the original experiments that identified quinacrine’s antiprion properties [Doh-Ura 2000, Korth 2001], the first compound library screen against prions [Kocisko 2003], and most subsequent major drug discovery efforts in multiple laboratories worldwide [Kawasaki 2007, Ghaemmaghami 2010, Wagner 2013].
The Prusiner lab used quinacrine as a positive control this assay for years [Ghaemmaghami 2010, Poncet-Montange 2011, Silber 2013]. Using this assay, they discovered the 2-aminothiazole lead [Ghaemmaghami 2010], which in turn led eventually to the development of IND24, which can extend survival time by ~60% in RML-infected mice [Berry 2013], and by >100% in CWD-infected mice [David Berry’s platform talk at Prion2015], though it is unfortunately ineffective against human CJD prions [Berry 2013]. Kurt Giles has expressed curiosity at the fact that, so far, all of the leads from these screens that they’ve tested in vivo against RML prions actually turn out to work. In other words, they haven’t found a second quinacrine, a second molecule that works beautifully in cells but not at all in vivo.
So how can it be that quinacrine is the most potent antiprion compound in cell culture, yet wholly ineffective in vivo?
When people searched for an explanation for quinacrine’s failure in vivo, attention quickly turned to its pharmacokinetics. Early reports differed as to whether quinacrine did or did not reach therapeutic concentrations in the brain in sheep [Gayrard 2005] and in mice [Yung 2004]. Prusiner and collaborators studied the issue extensively [Huang 2006, Ahn 2012] and found that quinacrine is exported from the brain by P-glycoprotein (encoded by ABCB1 in humans), a protein also known as MDR1 for its role in multiple drug resistance in cancer [e.g. Trock 1997, Patch 2015]. They therefore obtained Mdr1 knockout mice and found that in these mice, oral treatment with 40 mg/kg/day of quinacrine was sufficient to sustain 100 μM concentrations of quinacrine in the brain, about 2.5 orders of magnitude above its cellular EC50 [Ghaemmaghami & Ahn 2009]. Yet even in these mice, continuous treatment with quinacrine produced no survival benefit. Instead, across a battery of different dosing regimens, the only significant increases in survival time were seen when the mice were treated only transiently with quinacrine. The largest (and still slight, but significant) effect was seen when mice were dosed for only 10 or 20 days, extending the survival time from 127 days to 141 days (+13%, p < .01) [Table 1].
Indeed, when those mice were sacrificed at various timepoints, the PK-resistant PrPSc in their brains was found to decline only transiently after the initiation of quinacrine treatment, before recovering after about 25 days [Figure 3]. This finding was mirrored, albeit at a different time scale, in non-dividing cell cultures, where PK-resistant PrPSc reached its nadir after 2 days of quinacrine treatment, and fully recovered by about 5 days [Figure 4]. Prions from the brains of quinacrine-treated mice showed a slightly different signature in conformation-dependent immunoassay — it took slightly less GdnHCl to denature the prions enough to expose the D18 epitope — which was interpreted to mean that their conformation had changed [Table 2]. This became the first story of prions developing drug resistance through conformational change, a phenomenon which has since been observed for other compounds both in cell culture [Li 2010] and in vivo [Berry 2013].
But the drug resistance phenomenon seems inadequate to entirely explain quinacrine’s lack of in vivo efficacy. After all, IND24 also engenders drug resistance, at least in RML prions, yet it does still extend survival in vivo [Berry 2013]. And here’s an interesting tidbit: Kurt Giles’ Prion2015 poster demonstrated that IND24 is actually more effective at an intermediate dose rather than at its highest tolerated dose, and that it is more effective upon intermittent rather than continuous dosing. One possible interpretation of these data would be that IND24 is at its best not when it obliterates all of the IND24-sensitive prions, but rather, in a delicate balance where the drug-sensitive strain and the drug-resistant strain are kept in competition with one another.
If that’s true, then one might wonder whether the problem with quinacrine is actually that it is too potent. But that would surely be nonsense. If the issue with quinacrine were simply that it was too potent, then one would have expected that it would work better in wild-type mice, where its brain concentration is 5-10x lower, compared to Mdr1 knockout mice [Huang 2006]. Instead, under the dosing regimens where quinacrine does have a marginal effect on survival, that effect is pretty comparable between wild-type and Mdr1 knockout mice [Ghaemmaghami & Ahn 2009, Table 1]. And for what it’s worth, the problem is not limited to quinacrine. Two other quinoline compounds, amodiaquine and mefloquine, which are reported to have similar EC50 values of ~500 nM [Kocisko 2003, Kocisko & Caughey 2006], are likewise ineffective in mice [Kocisko 2004, Kocisko & Caughey 2006].
So although it’s hard to ever rule out one of these explanations, it seems quinacrine’s lack of in vivo efficacy cannot be trivially explained away by pharmacokinetics nor drug resistance nor a paradoxical effect of its potency. After discovering that quinacrine does not reduce PK-resistant PrPSc levels in the long run in non-dividing cells, the Prusiner lab briefly put forth a hypothesis that only compounds that do have this activity in non-dividing cells would be effective in vivo, and then immediately disproved this hypothesis by showing that cpd-b, which does work in vivo, more than doubling survival time [Kawasaki 2007, Lu & Giles 2013], doesn’t work in non-dividing cells either [Silber 2013].
At the end of the day, as Emiliano Biasini has pointed out to me, it’s hard to speculate as to why a compound does or does not work when we don’t know its mechanism of action.
What is quinacrine’s mechanism, then? I found four different proposals in the literature:
- Binding PrPC. One group reported NMR data suggesting an interaction between quinacrine and residues 225-227 (YYQ) of recombinant human PrP [Vogtherr 2003]. The experiments were done at 5 mM quinacrine, a concentration fully 10,000 times higher than the cellular EC50. The authors argued that this mechanism could nevertheless plausibly explain quinacrine’s cellular activity if the drug is highly concentrated in mammalian lysosomes (as observed in malaria parasites’ food vacuoles). The amino acid sequence that quinacrine was shown to interact with here (YYQ) differs in mouse PrP (where it is YYD, see here), and the cellular antiprion effects have been observed for mouse PrP. In other experiments, at lower concentrations of 100 μM or 40 μM, quinacrine appeared to have almost negligible affinity for PrPC as measured by surface plasmon resonance - more than an order of magnitude lower than the signal observed for Congo red [Kawatake 2006, Touil 2006].
- Binding PrPSc. A solid-phase matrix functionalized with quinacrine was found to pull down PK-resistant PrPSc from prion-infected mouse brains, while exhibiting undetectable affinity for PrPC from uninfected mouse brains [Phuan 2007]. The pulldown could reproduced after digestion with proteinase K (which would be expected to digest most other proteins) and after digestion with benzonase (which degrades nucleic acids), suggesting that quinacrine was binding directly to PrPSc without any intermediary. One caveat is that recovery was reported to be “low”, with only an estimated 4.8% of PrPSc being captured.
- Redistributing cholesterol. PrPC localizes to lipid rafts on the plasma membrane [Gorodinsky & Harris 1995], and these appear to be important for prion conversion, as lovastatin reduces PK-resistant PrPSc accumulation in ScN2a cells with an EC50 of 300-500 nM [Taraboulos 1995, Kocisko 2003]. One study reported that three other compounds known to redistribute cholesterol (U18666A, amiodarone, and progesterone) also possess antiprion activity, and that staining of quinacrine-treated ScN2a cells showed a migration of cholesterol from the cell surface to internal compartments [Klingenstein 2006b].
- Covalent modification of PrP. One study reported that quinacrine and related compounds transfer acridin-9-yl groups to lysine residues in PrP [Sebestik 2006].
Of these proposals, binding PrPSc seems attractive for its ability to potentially explain the development of drug resistance: prions change conformation, abolishing the quinacrine binding site. If true, one might expect that quinacrine’s binding site may also be specific to RML prions, and absent from other strains of prions. The evidence from other prion strains, however, only adds to the confusion. One study which found quinacrine to have an EC50 of 230 nM against RML prions also reported slightly higher EC50s of 590 nM against 22L prions and 1.88 μM against Fukuoka-1 prions [Nguyen 2011]. It’s hard to know what to make of that information, as different N2a cell clones give different EC50 values even with the same strain [Kawasaki 2007]. But in that same study, a new molecule they synthesized, referred to as Compound 3 (structure shown further below), had an EC50 of 35 nM against RML prions but no detectable activity against 22L or Fukuoka-1 up to the toxic concentration of 5 μM [Nguyen 2011]. If we assume that Compound 3 has the same mechanism as quinacrine, then this seems to support the idea of the target being a binding pocket whose shape is altered on different prion strains. Equally striking, quinacrine actually increases the level of proteinase K-resistant PrPSc in cells infected with CWD prions [Bian 2014]. In interpreting all of these results, remember that across all prion strains studied, PK-resistant PrPSc is the cellular readout, so we actually have little knowledge of what happens to PK-sensitive PrPSc, or to infectious titer, upon treatment with quinacrine.
Arguably, the observation that quinacrine has different effects on different prion strains might be consistent with the hypothesis that quinacrine binds PrPSc. If PrPSc really is the direct target of quinacrine, though, then there is much that we must simply write off as coincidence. For instance, quinacrine also inhibits [PSI+] prions in yeast [Bach 2003]. Granted, it is not unheard-of for a molecule to bind to amyloids composed of different substrate proteins - for instance, Congo red and ThT both bind to diverse types of amyloids [reviewed in Sipe & Cohen 2000], and certain polythiophenes will bind to amyloids composed of IgG light chain, serum amyloid A, or transthyretin [Nilsson 2010]. But could there really be a quinacrine-binding structural element shared between [PSI+] and RML prions yet not shared between RML prions and quinacrine-resistant RML prions?
And then there is the apparent coincidence of quinacrine and the quinoline class’s dual antimalarial and antiprion properties. These properties might be separable — methylene blue, which has antimalarial activity, seems to lack antiprion activity, although toxicity precluded testing it at concentrations above 1 μM [Korth 2001]. Yet all seven FDA-approved quinoline ring system-containing antimalarials (top) do inhibit RML prions in culture, and one more extensive study of further analogs has suggested shared a structure-activity relationship for antimalarial and antiprion activity [Klingenstein 2006a]. All of the data from that study are only presented in the form of tables, so I have plotted them (data, code):
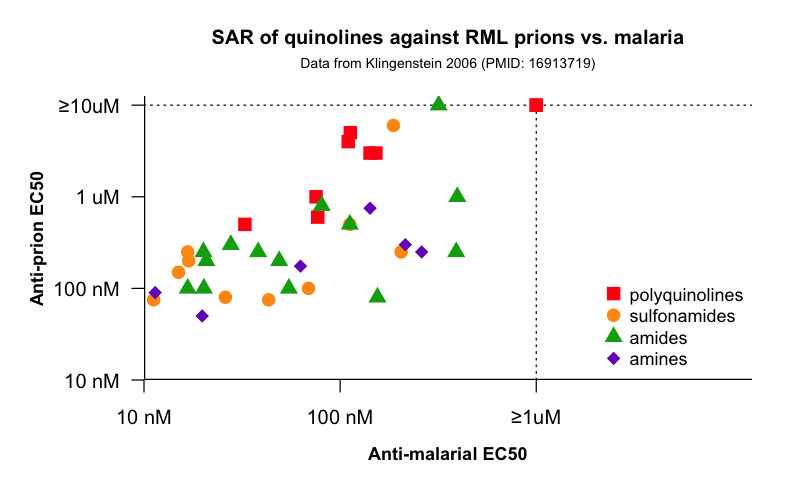
According to my calculations (which appear to differ slightly from what is reported in the paper, though the overall conclusion is the same; you can see my code) the overall Spearman’s correlation between the antimalarial and antiprion EC50s is ρ = .68 (p = 1.3 × 10-6) and is strongest within the polyquinoline class, indicated by red squares. And notably, there are no dots in the upper left or bottom right corners - no compounds that strongly inhibit one pathogen and don’t at all inhibit the other. This seems like evidence for a shared target, and the authors certainly assert so: “antiprion SAR in ScN2a cells were similar to antimalarial SAR in a cell model of malaria… suggesting that some molecular targets of antiprion and antimalarial substances overlap” [Klingenstein 2006a]. But obviously, Plasmodium falciparum don’t have PrP, so it would be hard to reconcile this with the notion that quinacrine directly binds PrPSc in prion-infected mouse cells.
Several indepepndent groups have undertaken SAR studies to make a better analogue of quinacrine, swapping in different functional groups and/or piling on multivalent combinations [May 2003, Murakami-Kubo 2004, Klingenstein 2006a, Nguyen 2011,Mays 2012]. These studies have collectively revealed that, in cell culture, it is indeed possible to do better than quinacrine. While it’s tricky to compare EC50s between studies, the reported values for these few examples shown below are all about 1-2 orders of magnitude smaller than for quinacrine, so it seems safe to say they are more potent:
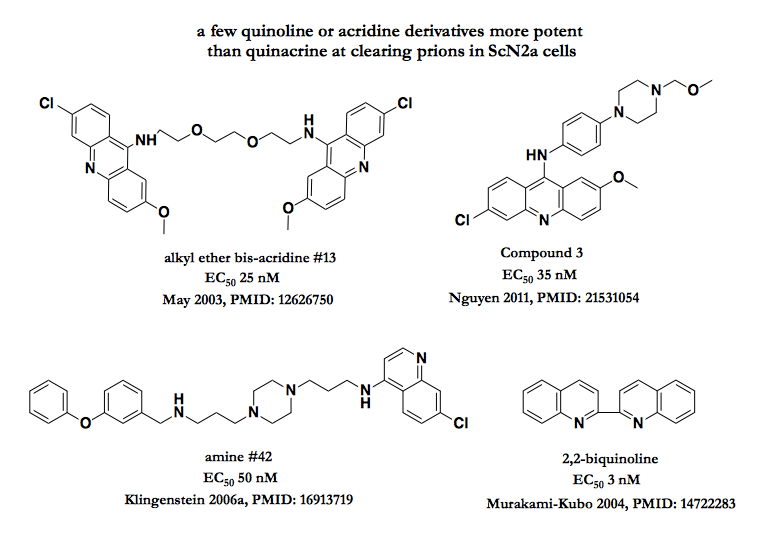
Sometimes you win by making the molecule more complicated, and sometimes you win by making it simpler. Look at 2,2-biquinoline! What is that thing?! How can that be 100 times more potent than quinacrine? And what does it tell us about the mechanism of action of this whole class? In conversations with chemists at the Broad Institute over the past few weeks, I’ve heard people react to very simple molecules with skepticism, wondering how something so simple could confer sufficient specificity to a target of interest to be viable as a drug. Yet one of these same scientists has pointed out that we are all humbled by the success of dimethyl fumarate (molecular weight: 144), which, marketed as Tecfidera®, has become a blockbuster drug for multiple sclerosis, generating almost $1 billion per quarter in revenue. So perhaps 2,2-biquinoline can’t be written off just for its simplicity — but like quinacrine, it appears to be completely ineffective in vivo [Murakami-Kubo 2004].
All told, four members of this quinoline class have been tested in vivo: quinacrine, 2,2-biquinoline, amodiaquine, and mefloquine [Collins 2002, Doh-Ura 2004, Murakami-Kubo 2004, Kocisko 2004, Kocisko & Caughey 2006], and none affected survival. You can’t prove that no molecule in this class would work, but their collective failure so far, together with the discovery of other leads that do turn out to extend survival in vivo [Kawasaki 2007, Berry 2013, Wagner 2013], probably means that the quinolines are no longer the lowest-hanging fruit for trying to find a drug for prion disease. Still, the sheer bizarreness of their whole story, as told here, makes me continue to wonder whether their mechanism of action might hold some fundamental insight about the biology of prions.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.