New high-throughput screening hits from the Prusiner lab
I just learned that the Prusiner lab has published the results of two new high-throughput screens for antiprion compounds, with a number of promising leads [Silber 2013, U.S. Patent Application WO2013033037A2]. Here’s what’s in this new paper.
dividing vs. stationary cell cultures
The paper describes two primary screens and a great deal of followup work on the hits from these. The method was an ELISA to detect proteinase K-resistant PrPSc, essentially the same as in the Prusiner lab’s previous efforts [Ghaemmaghami 2010, Poncet-Montange 2011] and as reviewed in this post.
The main difference from previous screens that they did one screen on continuously dividing ScN2a cells (actually, a subclone called ScN2a-cl3) and one screen on “stationary” ScN2a cells. ”Stationary” here almost means “non-dividing,” but not quite. When I Googled this term I found definitions suggesting that “stationary” means simply that the number of cells is not increasing – some might still be dividing, but at no greater a rate than others are dying off. N2a cells have been in continuous culture for decades and have been selected, no doubt, for their propensity to grow, so perhaps completely arresting cell division is a challenge and maybe that’s why no one is claiming to study “non-dividing” cells. In practice, the “stationary” cultures are simply those to which sodium butyrate was added to inhibit cellular proliferation. As far as I can tell, the intent here is to study something slightly closer to the in vivo situation, where prions kill post-mitotic cells (neurons).
The backstory is that quinacrine, which eventually proved to have no therapeutic activity in mice, reduced PK-resistant PrPSc in dividing cells [Doh-Ura 2000, Korth 2001] but did so only very transiently in stationary cells (again, arrested with sodium butyrate) [Ghaemmaghami & Ahn 2009], apparently because prions rapidly developed resistance to it. It was therefore hypothesized that maybe compounds are more likely to be effective in vivo if they’re effective in stationary cells [Ghaemmaghami & Ahn 2009]. This new study was certainly designed with that hypothesis in mind, and it even tests the hypothesis a tiny bit – more on that in a moment.
54,250 compounds were screened in dividing cells and 50,850 (mostly the same compounds I think) in stationary cell cultures. The hit rate was ten times higher in dividing cells. 5.9% of compounds reduced PrPSc by at least 30% in dividing cells, but only 0.65% did so in stationary cells. In the discussion, the authors offer one possible explanation for this. In dividing cell cultures, PrPSc accumulation is determined by 1) PrPSc formation, 2) cell division, which dilutes PrPSc, and 3) PrPSc breakdown [Ghaemmaghami 2007]. Each of these three factors is one possible mechanism by which drugs could act. In stationary cells, the authors argue, perhaps only mechanism #3 is possible to detect.
As for mechanism #2, compounds that promote cell division might not directly reduce PrPSc, but they dilute it and thus make it easier to digest – or in other words, they increase the number of cells capable of degrading PrPSc. If there are compounds in these chemical libraries that promote cell division, those would probably not show up as hits in stationary cells, and that’s probably a good thing, since those compounds wouldn’t actually be of any use in vivo.
Mechanism #1 is a bit harder to understand, and the paper doesn’t explain exactly why they don’t think mechanism #1 would be detected in stationary cells. What I’m guessing is that the change in PK-resistant PrPSc over the 5 days that the cells were incubated during the screen is just not a very large increase over its starting level if the cells are not dividing. Say the cells start with 1.0x rPrPSc and at the end of 5 days they have 1.2x rPrPSc. If that’s true, then even compounds that completely abolish rPrPSc formation would only be perceived as a 1-1.0/1.2 = 17% reduction in rPrPSc compared to negative controls, and wouldn’t be hits in the screen. And that’s a bad thing, because such compounds might very well be effective in vivo, since prion disease, quick though it may be, does last longer than 5 days.
None of this so far actually tests the hypothesis that in vivo activity is associated with activity in stationary cells. Though far from an exhaustive test of this hypothesis, the authors did look at Compound B, which is effective in vivo [Kawasaki 2007, Lu & Giles 2013] – in fact, it’s about as effective as IND24 and anle138b against RML prions – and found that it had no effect in stationary cells. Though this doesn’t disprove that compounds effective in stationary cells might be somewhat more likely, on average, to be effective in vivo, it at least shows it’s not an absolute requirement.
cheminformatics
I read closely all the sections on informatics in the paper to see how they analyzed their data. It was almost all out-of-the-box closed source software – they used SARvision for SAR, Qikprop for predicting physicochemical properties (including blood brain barrier permeability), and Collaborative Drug Discovery for managing the data.
The one pretty interesting thing that was implemented specifically for this study was how they selected the compounds to screen. UCSF’s Small Molecule Discovery Center has > 180,000 compounds available for screening, and the authors wanted to pick the best possible subset of 54,000. They wanted to maximize the “predicted bioactivity” of the individual compounds while also maximizing the chemical diversity of the whole subset, and with the catch that the compounds were already in 96-well plates, so they had to choose plates, not compounds.
The solution: they gave each compound a score for predicted bioactivity based on ChEMBL E values, and then ranked them in order of increasing molecular weight and gave each compound a point if its Daylight fingerprints differed from all previous compounds by a Tanimoto coefficient of at least .7, using SUBSET, and then picked the plates with the highest total score. I still need to learn more about what each of these things means.
screening results
The cutoff for a “hit” was a reduction in PrPSc of at least 30% in dividing cells. Based on this cutoff, 2100 compounds were re-screened to confirm the original finding and tested for cell viability in a calcein assay (calcein is blue-green dye that only living cells incorporate), to make sure they weren’t just killing the cells. 970 compounds passed these tests, and based on the SAR these were about 20 distinct chemical structures, of which 14 looked promising (the others had properties that made them potentially undruglike). They they picked 2 compounds from each of the 14 lead categories and did dose-response testing (by both ELISA and Western blot) to find the EC50, the dose at which they have half of their maximum possible inhibitory value. For a drug, it’s great to have an EC50 in the picomolar range, though if you’re just looking at lead compounds you have yet to optimize, then nano- or micro-molar is a viable place to start. All of the 14 leads had some compounds in the 1-10 μM range, and some had compounds with EC50 < 1 μM. I suspect that these EC50 values must have been the data that Dr. Prusiner flashed on the screen as a barplot during his speech at Prion2013.
Based on the 14 leads, they were able to order 467 similar compounds to be able to do SAR and figure out which properties were associated with the strongest antiprion activity. But interestingly, some of the broadest observations seemed to come not out of SAR per se but out of examining the top hits visually:
A conjugated aromatic or heteroaromatic ring system was prominent in all lead structures. This ring system is comprised of more than two aryl or heteroaryl groups joined in a fused or linear fashion. In the case of linear aromatic systems, two aromatic rings can be linked directly via a carbon-carbon bond or a linker such as an amide or double bond. Further analysis revealed that compounds with better potency (<1 μM) were from leads having core structures possessing a flat coplanar or near coplanar conformation…
So in short: the best antiprion compounds are flat, linear and full of rings. Sound familiar? That’s a good description of IND24, cpd-b and anle138b, the only three small molecules to show a strong effect in vivo so far. That the new hits from this much larger high-throughput screening effort follow the same pattern may suggest a common mechanism.
Finally, the paper also covers in vivo pharmacokinetics for 16 compounds. The authors found 4 compounds that had good bioavailability and blood brain barrier permeability. 2 of these compounds (IND45193 and IND52851) reached a maximum concentration > 1 μM in the brain after an oral dose of 10 mg (in mice), which is about about equal to their EC50s, suggesting that these compounds are already capable of reaching therapeutic concentrations after oral delivery – and that’s before much lead optimization has been done. I’ll venture a guess that these compounds, or analogues thereof, might turn out to be the next compounds tested for in vivo efficacy in the Prusiner lab.
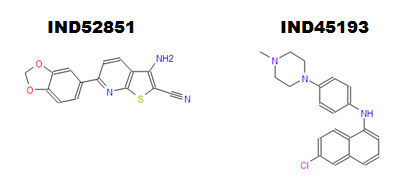
IND52851 is a thienopyridine and IND45193 is a quinoline. SMILES are Nc3c(C#N)sc4nc(c2ccc1OCOc1c2)ccc34 and CN4CCN(c3ccc(Nc1cccc2cc(Cl)ccc12)cc3)CC4 respectively. It caught my eye that IND52851 contains a methylenedioxyphenyl group like anle138b.
aside: FDA-approved drugs
Among the compounds screened in this study were 1420 FDA-approved drugs, and the authors report specifically on the 15 of them that appeared to have antiprion activity in stationary cells. They are: dextran, Congo red, carvedilol, tetrandine, ethoxazine, dihydroergotamine, acepromazine, amlodipine, fendiline, tamoxifen, desloratidine, apomorphine, amiodarone, hexadimethrine, and enoxaparin.
What amazes me is that there is no overlap here with the set of FDA-approved drugs reported to have antiprion activity in any of three previous screens [Kocisko 2003, Poncet-Montange 2011, Karapetyan & Sferrazza 2013]. As far as I know, of these 15, only dextran and Congo red were previously reported to have antiprion activity [Ehlers & Diringer 1984, Caughey & Race 1992] and those were the two strongest hits.
discussion
This paper seems to represent a pretty enormous amount of work. It covers everything from the screening of compound libraries to in vivo pharmacokinetics, and given the lag in time from when scientific research is done to when it appears in print, it seems very likely that by now some of these compounds are already being tested in prion-infected mice to see if they extend survival. Compared to the screens I discussed in my last post, these efforts seem to be a lot further down the path towards finding a compound that works in vivo. That’s exciting news.
One issue that’s not explicitly addressed in this paper is how to handle the potential drug resistance and strain specificity problems revealed by the Prusiner lab’s 2-aminothiazole work published last month [Berry 2013]. Here, as in previous screens, chemicals are screened against RML prion-infected N2a cells, and undergo a great deal of SAR, pharmacokinetics, and (presumably) survival testing in RML-infected animals before they ever see a human prion. The screen against stationary cells here seems designed to partially address this problem: if compounds are effective in stationary cells, that must mean that at least it is difficult to prions to evolve resistance to them, and that might be a good sign that the compound inhibits prions of a variety of different conformations. Sadly, this paper reveals some limitations of this approach: the hit rate was much lower in stationary cells than dividing cells, and compounds that inhibit PrPSc formation might not show up as hits in stationary cells at all. A different approach which has been floated is to screen against quinacrine- or IND24-resistant RML prions, and I’ll be curious to see if that approach surfaces in future papers from the Prusiner lab.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.