Native function update
So what’s the native function of this protein?
At every talk about prion disease that I either give or attend, without fail, someone in the audience will ask this question. It’s a question that I, and many other scientists, stumble over. A decent short answer is “we don’t know,” but that’s not quite completely true. And now, a new study out last week from the Aguzzi lab [Kuffer & Lakkaraju 2016] has brought us one step closer to having a real answer to this question.
In short: the authors have determined that a G protein-coupled receptor (GPCR) called Adgrg6, which was previously known to be expressed on the surface of Scwhann cells and required for myelin formation, acts as a receptor for the N terminus of PrP, mediating PrP’s role in myelin maintenance on peripheral nerves.
This blog post is a quick look at how we got here, and what comes next.
the sordid history of PrP knockout phenotypes
Prion diseases are caused by a gain of function. We know this because, among other observations, PrP overepxression accelerates disease, PrP knockout confers resistance to disease, and the phenotype of prion disease is entirely different from that of PrP knockout [Bueler 1993, Fischer 1996]. Here’s a backgrounder.
Most studies trying to decipher PrP’s native function have looked at cells or animals lacking PrP. The first line of PrP knockout mice, known as Zürich-I or ZH1, were first created and characterized 24 years ago [Bueler 1992]. Their most salient phenotype was, of course, invincibility against prion infection [Bueler 1993], but obviously, PrP didn’t evolve to render us susceptible to a rare and horrifying disease, so it surely has to be doing something else as well. But PrP knockout mice seem fine: they’re viable, fertile, have normal lifespans, and after a battery of tests, their creators couldn’t find a darn thing wrong with them [Bueler 1992].
In the years since then, we’ve accumulated more lines of PrP knockout mice — there were 7 at last count: Zürich-I, II, and III, Nagasaki, Edinburgh, GFP, and Riken [Nuvolone & Hermann 2016]. We’ve also accumulated a bewildering number of claims of different PrP knockout phenotypes — 26 the last time anyone did a systematic review [Steele 2007, see Table 1].
But a great many of the reported phenotypes have either failed replication or been flat-out refuted. For instance, the first reported knockout phenotype was a difference in electrophysiological properties of mouse brain hippocampal slices [Collinge 1994], which the Prusiner lab found that it couldn’t replicate [Lledo 1996]. After it was discovered that PrP’s octapeptide repeats bind copper [Hornshaw 1995], there were claims that PrP possessed (or somehow otherwise enhanced) superoxide dismutase activity [Brown 1997], but this was spectacularly disproven by a series of in vivo experiments showing that various levels of PrP expression, in the presence of various levels of SOD1 expression, had no effect on total superoxide dismutase activity in the brain [Hutter 2003].
Most of the time, we never find out why a report of a PrP knockout phenotype got it wrong — was it bias, error, or an experimental confounder? But in just a few cases, the root cause has actually been identified. Most famously, as told in detail in this post, the Nagasaki line of PrP knockout mice exhibited a late-onset cerebellar neurodegeneration phenotype which was later shown to be due to ectopic expression of the downstream Prnd (encoding Doppel or Dpl) as a result of the gene targeting strategy used to knock out PrP. In a more recent example, it was claimed that PrP knockout macrophages exhibit enhanced phagocytic activity [de Almeida 2005], but the Aguzzi lab showed that this phenotype was actually attributable to functional polymorphisms in the Sirpa gene between the B6 and 129 mouse lineages [Nuvolone 2013]. The Zürich-I PrP knockout mouse was originally created on a 129 background, and then backcrossed to an almost pure B6 background, but it inevitably retained some 129 sequence around the PrP gene. Sirpa is located 2.2Mb upstream of Prnp on mouse chromosome 2, sufficiently tight linkage that PrP knockout mice, even after generations of backcrossing, had still retained the more functional 129 Sirpa haplotype rather than the less functional B6 Sirpa haplotype. This was further validated by the fact that the Zürich-III mouse, which is pure B6 and has only a TALEN-induced frameshift in Prnp, does not have the phagocytosis phenotype [Nuvolone & Hermann 2016]. Genetic linkage confounders make it critical to know which knockout mouse line(s) were used, and on what genetic background, in order to interpret any claim of a PrP knockout phenotype. Yet, amazingly, such details are often buried in a Methods section.
Along with all of the questionable results, there have been a few findings that have stood out as having a somewhat stronger evidence behind them.
One is the claim that, when researchers electrically cauterize a cerebral artery in order to induce a stroke, PrP knockout mice end up having larger infarct volume than wild-type mice do. This was first observed only in the Edinburgh knockouts [McLennan 2004], but later replicated in the Zürich-I and Nagasaki lines [Sakurai-Yamashita 2005, Weise 2006] and found to be rescued by the Tga20 full-length PrP transgene array [Spudich 2005]. These various findings came from a few different research groups, with no other groups refuting it, so this might be real. If so, we don’t have any molecular resolution on how it works at present.
Another claim which looked possibly credible was the report that PrP knockout mice had a defect in peripheral myelin [Nishida 1999]. I don’t have full text access, but apparently this phenotype was observed in both Zürich-I and Nagasaki mice and was rescued by a full-length PrP transgene. The peripheral myelin pathology in those mice looked similar to pathology seen in some mice expressing PrP transgenes with deletions, which prompted the Aguzzi lab to take a much closer look.
PrP’s signaling role in peripheral myelin maintenance
Here’s what we learned from the exceptionally thorough battery of experiments they reported more than a decade later [Bremer 2010]. PrP knockout mice have a defect in the maintenance of myelin on peripheral nerves, described as a chronic demyelinating polyneuropathy or CDP. This phenotype was replicated across three knockout lines (Zürich-I, GFP, and Edinburgh [Bremer 2010]) and later a fourth (Zürich-III, [Nuvolone & Hermann 2016]), and persisted on four genetic backgrounds (B6, 129, Balb/c, and mixed), so it’s definitely real. It’s progressive: the mice seem to be born with normal myelin, but have some deficits visible by 10 weeks of age, and these deficits are more pronounced by 60 weeks of age. It’s also mild, in the sense that although the changes are readily observable by histology and can also be detected by electrophysiological measurements, they barely scrape the surface of a behavioral phenotype. At 60 weeks, only a modest difference in grip strength and time to jump off of a hot plate was observed, and even then, the difference was not replicated across all experiments.
The way myelination works in the periphery is that Schwann cells, a type of glia, physically envelop the axons of neurons and wrap them in myelin [reviewed in Salzer 2015], so the authors next asked whether PrP expressed on neurons or on Schwann cells was required for myelin maintenance. The demyelination phenotype could be rescued by PrP expression restricted to neurons, but not by PrP expression restricted to Schwann cells, meaning that PrP has to signal in trans from neurons to Schwann cells. Of the several mutant transgenes tested, only those competent to undergo alpha cleavage could rescue it, suggesting that perhaps the N1 fragment released by that cleavage event (the flexible tail or FT in Aguzzi lab parlance) was responsible for the signaling.
The new study out last week confirms this hypothesis, identifying a receptor for the flexible tail or N1 PrP fragment: a G protein-coupled receptor (GPCR) known as Adgrg6, which is expressed on the surface of Schwann cells [Kuffer & Lakkaraju 2016].
Before getting into the details of the experiments, here’s what I learned in terms of background on Adgrg6. It belongs to a subclass of GPCRs called adhesion GPCRs and was only renamed (last year); its former name of Gpr126 can still be seen in many papers (and ExAC). The earliest references to Adgrg6 appear to be from papers simply mining the human genome for things that look like a GPCR [Fredriksson 2003, Bjarnadottir 2004], but nothing specific was known about the gene or protein at that time. A random mutagenesis screen in zebrafish identified several mutants with defects in myelin formation [Pogoda 2006], two of which, st49 and st63, eventually turned out to be nonsense and missense mutations (respectively) in the gene encoding the zebrafish ortholog of Adgrg6 [Monk 2009]. The Adgrg6 knockout mouse proved to have a similar phenotype [Monk 2011]. The phenotype of ADGRG6 knockout in humans is incredibly severe: last year, a study identified three families in which homozygous missense or protein-truncating variants in this gene had resulted in perinatal lethality within 1 hour of birth due to a complete lack of myelination, with no myelin basic protein on peripheral nerves at all [Ravenscroft 2015]. Aside from the very severe knockout phenotype, ADGRG6 is also a GWAS hit for adolescent idiopathic scoliosis (a condition involving curvature of the spine), with a modest odds ratio of 1.28 [Kou 2013]. The extracellular domain of Adgrg6 required for GPCR activation has been identified [Liebscher 2014], and ligands have been identified including collagen IV [Paavola 2014] and laminin-211 [Petersen 2015]. You can see in GTEx that ADGRG6 mRNA is expressed in peripheral nerves but not brain. (ProteinAtlas is also mostly negative for expression in brain, although they do claim some expression of protein in neuropil and endothelial cells in cerebral cortex with one antibody).
Taking all this into account, Adgrg6 sounds like a good candidate to be PrP’s receptor: it is a GPCR with a known role relating to peripheral myelin. On the flipside, Adgrg6 might seem like an imperfect match in the sense that the PrP knockout phenotype is progressive mild, whereas the Adgrg6 knockout phenotype is perinatal lethal. The former consideration seems to be the hypothesis on which the experiments reported here were conducted: they never tested an exhaustive library of GPCRs, instead focusing primarily on Adgrg6, using just a handful of other GPCRs as controls. But at the end of the paper they also return to the latter issue, and provide a potential explanation for the discrepancy.
Here’s a quick summary of the experimental workflow. First, they confirmed by immunostaining that N-terminal fragments of PrP do indeed bind to the surface of Schwann cells, as hypothesized in [Bremer 2010] and as required if N1 is indeed a signaling molecule. Second, they asked whether N-terminal fragments bound to Schwann cells caused a spike in cAMP signaling in those cells. The answer was yes, and was confirmed both using recombinant PrP fragments and conditioned media from PrP-expressing cells. Accordingly, decreased overall cAMP levels were also observed in peripheral nerves from PrP knockout mice. At this point, since cAMP signaling is governed by GPCRs, they focus in on the hypothesis that Adgrg6 might be the receptor. They found that HEK cells expressing Adgrg6, but not any of several other GPCRs, responded to the N terminus of PrP with increased cAMP signaling, and conversely, Schwann cells with Adgrg6 deleted didn’t bind PrP nor exhibit any cAMP response to recombinant PrP fragments nor to conditioned media containing PrP N-terminal fragments.
Throughout the paper, they narrow down on which region of PrP is required for the signaling. By using different recombinant peptides and seeing which ones do or do not bind, and do or do not cause cAMP spikes, they narrow it down to PrP23-34 (KKRPKPGGWNTG). A scrambled version of this peptide was inactive, ruling out a non-specific ionic interaction with all those positively charged residues. Collagen IV, a known Adgrg6 ligand, turns out to have a similar sequence of GPRGKPG, so they further zoomed in on the corresponding segment of PrP, and found that mutating the lysines to alanines abolished PrP’s activity with respect to Adgrg6. Thus, the relevant segment of PrP may be just PrP23-29: KKRPKPG.
There were several other experiments I won’t go into in detail, such as examining transcription factors involved in myelination, and treating the original zebrafish mutants st63 and st49 with PrP peptides. Most interesting to me was the discussion of mice with Cre knockout of Adgrg6 specifically in Schwann cells starting from embryonic day 12 [Mogha 2013]. These mice were created a few years ago and were found to have a far milder phenotype than constitutive Adgrg6 knockout mice [Mogha 2013]. In the new paper, they compare these mice side-by-side with PrP knockout mice and conclude that their phenotypes are qualitatively similar. So apparently, initial myelin formation can be accomplished by Adgrg6 expressed either early on in embryonic development in Schwann cells (or later on in other cell types?) in the Cre mice, and can be accomplished via signaling from other Adgrg6 ligands in PrP knockout mice. Thus, both of these mouse lines have myelin that forms fine initially but deteriorates later on, whereas total absence of Adgrg6 keeps myelin from forming correctly in the first place.
Here’s my best attempt at a diagram depicting this whole model:
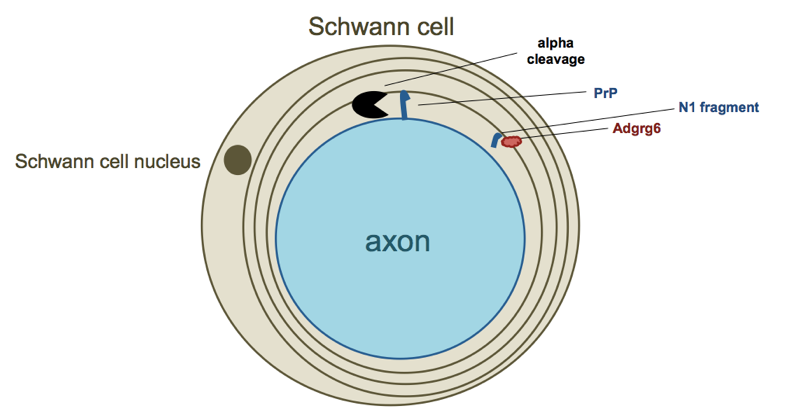
The process of sitting down to try to draw a diagram presented me with a lot of difficult choices. Is this whole signaling process taking place with immature Schwann cells that haven’t yet enveloped the axon, or with mature Schwann cells that have already engaged? I depicted the latter above, but I am not sure whether this is correct. Is the N1 fragment free floating through extracellular space to reach its target, or is it tightly confined within a sheath encircling the axon? I have depicted the latter, but again, I am not sure if that makes sense. To begin with, I don’t know much of anything about myelin biology, and then, even the experts may not yet have a clear picture of how exactly this works in terms of the exact timing and locations involved.
implications
As is typical for the Aguzzi lab, the experiments in this paper are remarkably thorough, examining the issue via several different approaches in several different model systems, including in vivo. Still, one can imagine some experiments that could be done to further test the model presented in this paper. The reported experiments suggest that the extreme N terminus — PrP23-29 or at most PrP23-34 — was the only region possess Adgrg6-stimulating effects. Therefore, the findings of this new paper predict that mice expressing PrPΔ23-88 on a knockout background [Supattapone 2001] should suffer from CDP just like PrP knockout mice do. The original paper reporting on CDP [Bremer 2010] examined the presence or absence of CDP in mice expressing six different PrP transgenes containing different N-terminal deletions. It found that only mice expressing PrP molecules that were incompetent to undergo alpha cleavage (required to release the flexible tail for its signaling role) had CDP, but it didn’t examine any mice that lacked the extreme N terminus (residues 23-32) of PrP. That seems like one important experiment to confirm the model presented here. Still, the results already reported look solid, so unless anyone shows otherwise, I’m going to assume that these findings are both correct and important.
The question that all of my colleagues who’ve seen this paper have asked me is, does this change anything about your therapeutic strategy? We’ll see what future discoveries reveal, but for now, my answer is a firm no.
First, Adgrg6 is not a drug target for prion disease. We don’t know what PrP’s gain of function in prion disease consists of, but it’s certainly not hyperactivation of Adgrg6, which isn’t even expressed in the brain. The authors agree, but speculate that inappropriate activation of other GPCRs that are expressed in the brain could be part of PrP’s gain of function. That’s a possibility, and it will be interesting to see what they find, but for now, the new findings don’t give us any specific new drug targets.
Second, none of the new findings dampen my enthusiasm for PrP-lowering therapies. Even if a therapeutic amounted to a total PrP knockout, everything we know about the PrP knockout phenotype is still incredibly mild compared with a prion disease phenotype. And in any event, it’s unlikely any drug could achieve a 100% reduction in gene dosage. A 50% knockdown is more realistic, even overly optimistic if anything. The peripheral myelin phenotype isn’t present in heterozygous PrP knockout mice [Bremer 2010], and the few heterozygous PrP knockout humans who we’ve managed to identify seem to be healthy [Minikel 2016], so it seems likely there exists a therapeutic window where ~50% knockdown of PrP could dramatically delay prion disease [Fischer 1996] while not resulting in any peripheral myelin pathology.
What’s next? This is probably not PrP’s only native function — PrP knockout mice do not have myelin pathology in the CNS [Bremer 2010], yet PrP is highly expressed in the CNS, so it may be serving some other function there. Indeed, PrP is pretty much expressed everywhere, so one has to wonder if it has functions in other cell types as well. The Aguzzi lab seems to be hard at work on solving these mysteries, and I look forward to the next update.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.