Prion2019 days 2-3 and poster session

We are in Edmonton, Alberta for the Prion2019 conference. These are my notes from a few of the platform talks on days 2 & 3 plus the poster session.
Christina Sigurdson: Modifying prion disease spread through the CNS

Dr. Sigurdson is interested in the role of co-factors in prion formation. In this talk, she examined the roles of heparan sulfate, PrP glycosylation, PrP GPI anchoring, PrP mutations, and ADAM10 shedding of PrP.
The inputs to the experiments largely revolved around four different prion strains — RML, ME7, 22L, and mouse-passaged CWD (mCWD) — being passaged into mouse models where these factors of interest are perturbed in different ways. For example, EXT1 is a gene involved in heparan sulfate biosynthesis, and in humans, it is associated with a dominant disease of haploinsufficiency known as hereditary multiple exostoses (OMIM 608177). As a tool she used Ext1+/- mice which are deficient in heparan sulfate biosynthesis [Forsberg & Kjellen 2001]. She also used GPI anchorless mice [Chesebro 2005], and another mouse model where PrP is GPI-anchored but missense mutations are used to abolish the N-linked glycans.
The several outputs from the experiments included things like measuring incubation times on 1st, 2nd, 3rd, and 4th passages to determine whether the perturbation created a species barrier (indicated by a progressive shortening of incubation time on repeated passage), as well as histology to characterize PrPSc plaques in the brain, and Western blots with the new antibody specific to ADAM10-shed mouse PrP [Linsenmeier 2018].
Heparan sulfate and other glycosaminoglycans (GAGs) have long been known to bind PrP and to be found in prion plaques in the brain [Snow 1989, Caughey 1994]. But their exact role in prion replication, and even whether they are friend or foe, has been rather confusing. For instance, treating ScN2a cells with exogenous heparan sulfate, or inhibiting endogenous heparan sulfate biosynthesis, are both reported to antagnonize PrPSc formation [Caughey 1994, Ben-Zaken 2003]. Sigurdson found that, at baseline in wild-type mice, heparan sulfate is found within PrPSc plaques in the brain in some mouse adapted prion strains, but not others, and that the different prion strains behaved very differently in the heparan sulfate-deficient mice. Similarly they found that the proportion of shed PrP in PrPSc plaques differed between strains, and the strains behaved very differently in anchorless mice.
A principal finding was that prion strains differed in their reliance on the various co-factors, and therefore, passaging into mouse models where these co-factors were perturbed had different results. When PrPSc was mostly GPI-anchored, there was not much heparan-sulfate bound, and these strains were not much affected by passage into mouse models lacking glycosylation or deficient in heparan sulfate. Other prion strains had PrPSc that was not-GPI-anchored, being instead comprised more of shed PrP, and in this case the strain bound heparan sulfate; for these strains, adding back the GPI anchor, glycosylation, or heparan sulfate production had a greater impact on strain properties. The presence or absence of a GPI-anchor was key to plaque or diffuse aggregate formation.
Joel Watts: Alpha-synuclein strains initiate distinct transmissable synucleinopathies

There is plenty of evidence that α-syn aggregates can induce templated protein misfolding, or in other words, can form “prions” [Luk 2012, Mougenot 2012, Watts 2013, Prusiner 2015], and there are various lines of evidence that α-syn can form different prion strains [Peelaerts 2015, Peng 2018, Woerman 2019], for instance, fibrils with different biochemical properties. But what hasn’t been shown yet is whether these “strains” are truly strains in the sense that they maintain their properties upon serial transmission in vivo. Today, Dr. Watts presented a project from his lab to generate conformationally distinct preparations of α-syn prions from recombinant human α-syn and then serially transmit them in TgM83(+/-) mice.
They used high and low salt conditions to create two different α-syn fibrils in vitro and did various characterizations to show that these were conformationally distinct. They then transmitted them to the TgM83(+/-) mice in three successive passages. Across all three passages, the high vs. low salt fibrils maintained several distinct properties including differences in incubation time, the nature of the clinical signs in the mice, pattern of α-syn deposition across different brain regions, morphology of aggregates on histology, localization of aggregates into different cell types (glia vs. neurons), and banding pattern on a gel after proteinase K digestion. Thus, these distinct α-syn preparations exhibit all the hallmarks of different strains.
These findings have been submitted for publication and are now in revision (Lau et al).
Stanley Prusiner: Arguments for Alzheimer’s and Parkinson’s diseases being caused by prions

Dr. Prusiner noted the enormous and growing public health burden of dementia, and reviewed the eight years he spent purifying infectivity from fractions of scrapie-infected rodent brain to identify the prion. He then recalled a pivotal moment of seeing visually that the green-red birefringent rods of PrP in prion disease appeared similar to those made of Aβ seen in Down syndrome brains. This led him to the speculation that Alzheimer’s was also a prion disease [Prusiner 1984]. He now believes that Aβ, tau, and α-syn all behave as prions in humans, and there are many other prions and functional amyloids in yeast and other species.
Dr. Prusiner presented a striking visualization of failure in Alzheimer’s drug discovery:
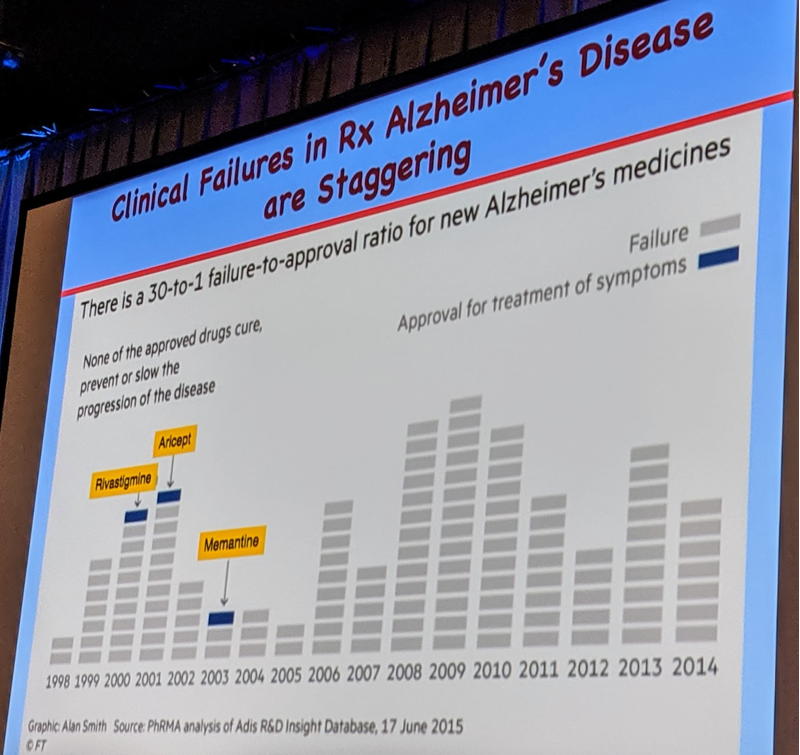
In short, in recent years there have been 30 failed Alzheimer’s drugs for every 1 approved drug, and the approved drugs have all been symptomatic treatments that have been very profitable but do not impact the underlying disease process. The recent failures have been particularly striking, as several anti-Aβ therapies that initially seemed rational and promising have failed. These include verubecestat (a small molecule BACE1 inhibitor), which if anything seemed to make people worse [Egan 2018, Egan 2019], and the anti-Aβ antibodies crenezumab, solanezumab [Honig 2018], and aducanumab. Prusiner believes that while Aβ and tau are the causal proteins in Alzheimer’s disease, one reason for the high rate of failure may be the focus on plaques, which may be a relatively biologically inert species, as opposed to focusing on prions, or in other words, the biological ability to conformationally template more protein.
Prusiner has therefore devoted much of his lab’s effort to developing better tools to study prions per se — ability to template protein conversion in cell culture assays, transgenic animals, and so on. To raise just a couple of examples, they can now measure tau prions using cells expressing tau-YFP [Woerman & Aoyagi 2016] and synuclein prions in cells expressing α-syn-YFP [Woerman 2015], and they can measure multiple system atrophy (MSA) synuclein prions in TgM83(+/-) mice [Watts 2013, Prusiner 2015]. They’ve used these sorts of tools to characterize and quantify tau, α-syn, and other prions in human brain samples. A recent finding is that among people with Alzheimer’s, the oldest individuals have higher total insoluble tau but lower tau prion activity [Aoyagi & Condello 2019].
A few observations about MSA. MSA is much more rapid than PD, with a disease duration of about 5 years, and often with orthostatic hypotension (blacking out when you stand up), which can be so severe that patients cannot get out of bed. Another striking feature is that MSA prions are very resistant to formalin treatment [Woerman 2018a], like PrP prions are. Bonafide MSA prions from human brain cannot replicate in cells with the SNCA E46K mutation (which causes Parkinson’s disease) [Woerman 2018b], consistent with a conformational difference between α-syn aggregates in PD versus MSA. However, α-syn fibrils formed in vitro do propagate in the presence of this mutation, which suggests their conformation is not the same as the conformation that occurs in vivo in human brain, and this makes Prusiner skeptical that these fibrils are a good model for drug discovery.
The Prusiner lab’s success in identifying compounds that inhibit RML prions, such as IND24, may hold lessons for drug discovery in non-PrP neurodegenerative diseases as well. He pointed to the finding that intermittent dosing of IND24 is more effective than continuous dosing, and that prophylactic dosing is more effective than later dosing [Giles 2015], and suggested these findings may point to variables that will need to be tested in transgenic mouse models for other neurodegenerative diseases in the development of therapies for those diseases.
Sonia Vallabh: Antisense oligonucleotides for the prevention of genetic prion disease

Sonia presented her work over the past five years to advance an antisense oligonucleotide (ASO) therapy for prion disease, and particularly for the prevention of genetic prion disease. This was work together with several collaborators including folks from Byron Caughey’s lab and Ionis Pharmaceuticals (pictured below) as well as McLaughlin Research Institute.

Me, Byron Caughey (collaborator on mouse studies), Anne Smith (clinical development at Ionis), Hien Zhao (preclinical development at Ionis), and Sonia Vallabh.
Sonia began by reminding us that she is a carrier of the PRNP D178N mutation and her personal goal in life is to prevent her own disease. She, like everyone here, hopes we can also achieve meaningful treatment for people who are already symptomatic, but for those who have predictive genetic testing, the highest good we can do is to intervene before onset and delay or prevent disease entirely, preserving full quality of life.
Sonia reviewed the evidence that lowering PrP is the right drug target in prion disease (a topic I’ve previously blogged here). PrP dose-dependently controls time to disease after prion infection in animals [Fischer 1996], the one knockout phenotype is relatively mild and not seen in heterozygous knockout mice nor humans with one inactivated allele of PRNP [Bremer 2010, Minikel 2016], and in the scheme of all human genes you could consider as drug targets, PRNP is among the most dispensible, appearing fully tolerant of heterozygous inactivation by loss-of-function mutations [Minikel 2019]. The question, then, is how can we realistically lower PrP in the human brain. She introduced ASOs and their mechanism of action and the data from ASOs currently in the clinic for spinal muscular atrophy, Huntington’s disease, and SOD1 ALS that suggest that ASOs can acheive good brain distribution, months-long duration of action, and perhaps 40% knockdown of a target protein, all with good tolerability. Our strategy is to develop a non-allele-specific ASO to reduce all PrP in the human brain, delivered intrathecally (through a lumbar puncture). The Prusiner lab previously partnered with Ionis to do preclinical experiments on one PrP-lowering ASO, and saw some efficacy, but the project was abandoned due to uncertainty about the mechanism of action of ASOs, poor tolerability of the one ASO tested in vivo in mice at 60 days-post infection, and the poor tolerability of intracerebral pumps that were used at that time for ASO delivery [Nazor Friberg 2012].
In 2014, Sonia and I approached Ionis and asked them to partner with us to re-start the development of ASOs for prion disease. Ionis identifed new ASOs that lower mouse Prnp RNA in the brain by about half. Sonia presented data from several efficacy experiments on these ASOs in prion-infected mice, at Byron Caughey’s lab and at our lab at Broad. With 2-3 doses beginning prophylactically, PrP-lowering ASOs delayed disease by about 60-100%, and findings replicated between the two labs. A scrambled control ASO was ineffective, showing that the mechanism of action is PrP RNA-lowering and not aptameric due to PrP binding. Efficacy was dose responsive — even just 20% knockdown of PrP at a low ASO dose was sufficient to confer measurable increase in survival and delay of symptoms, but more knockdown was better. Efficacy was observed against 5/5 mouse-adapted prion strains tested. We also tried dosing mice at 120 dpi, when prion neuropathology is established but the mice are not yet obviously symptomatic. At this timepoint, we identified two ASOs that were not well-tolerated, but two others that did increase survival time by at least half, again replicating between the Caughey lab and our lab. In a time-series experiment, where mice received chronic dosing of ASOs beginning at different timepoints, late treatment (105 - 120 dpi) resulted in a modest delay in the accumulation of prion disease symptoms (11-21%) while initiation of treatment earlier (anytime from 7 days before infection to 78 dpi) resulted in a more than two-fold delay, and experiments are still ongoing.
Sonia then shifted gears to thinking about the right strategy for testing a PrP-lowering therapy in the clinic. She noted that trials in symptomatic patients can’t tell us whether a drug might be able to prevent disease, especially since ASOs appear most effective when given earlier in disease in mice. Randomizing pre-symptomatic patients to disease onset is numerically impossible [Minikel 2018]. In Alzheimer’s disease, some trials are now pursuing “secondary prevention” — treatment of individuals with prodromal evidence of pathology but who are not yet symptomatic. But there does not appear to be a reliably detectable prodrome far in advance of prion disease onset that would facilitate such a paradigm in mutation carriers. Therefore Sonia has focused on a strategy for treating pre-symptomatic PRNP mutation carriers towards a biomarker endpoint measurable in healthy people before the disease process begins. Specifically, she has zeroed in on measurement of CSF PrP as a pharmacodynamic biomarker to read out the effect of PrP-lowering therapy. We met with FDA scientists in 2017 to ask if CSF PrP could be considered as a surrogate endpoint for Accelerated Approval of a PrP-lowering drug in healthy mutation carriers, and FDA was supportive and offered to work with us to assemble the necessary data to support this path.
CSF PrP appears to be CNS-derived, stable over time, and is reliably measurable provided that pre-analytical variability can be minimized, supporting its use as a pharmacodynamic biomarker [Vallabh 2019]. We launched a clinical research study at Mass General Hospital run by Dr. Steven Arnold that is recruiting healthy mutation carriers and controls to donate CSF for research. In samples collected so far, Sonia observes excellent short-term (2-4 month) within-individual test-retest reliability of CSF PrP across all mutations and controls, and in the limited longitudinal data, this stability appears to be maintained for at least a year. CSF total tau and NfL appeared normal in all mutation carriers, indistinguishable from controls and with no upward trend over time, and RT-QuIC was completely negative in the vast majority of carrier samples. These data suggest that cross-sectionally, if one recruits pre-symptomatic mutation carriers for research (and by implication for clinical trials), one will by and large find people who are not in a state of prodromal pathology. A prodrome, to the extent one exists in prion disease, may be too short or too rare to present a large enough population for trials. This supports the idea that a primary prevention approach based on lowering CSF PrP is a better shot on goal than a secondary prevention approach for clinical evaluation of a PrP-lowering therapy in pre-symptomatic people.
In conclusion, Sonia’s work has helped to advance a rational, targeted, PrP-lowering therapy for prion disease, and to lay out a potential path for clinical evaluation of such a therapy in pre-symptomatic mutation carriers based on a surrogate biomarker endpoint.
She’s my hero.
Luise Linsenmeier: Substrate-specific manipulation of the ADAM10-mediated shedding of PrPC

Shedding of PrP from the cell membrane by ADAM10 appears to play some protective role against prion disease [Altmeppen 2011, Altmeppen 2015]. Using a new antibody specific to shed mouse PrP [Linsenmeier 2018], Dr. Linsenmeier and colleagues have now set out to find approaches that can increase shedding and might lead to a therapeutic application. They figure that because ADAM10 cleaves a large number of different protein substrates, just increasing ADAM10’s activity globally may not be a viable approach; instead they need to increase its clevage specifically of PrP. They found that many (but not all) PrP antibodies increase PrP shedding in N2a cells, and this replicated in cerebellar organotypic cultured slices (COCS). They also identified an antibody that reduced shedding, as well as an antibody that reduced total PrP, apparently by causing rapid clustering, endocytosis, and degradation of PrP. Reported small molecule binders of PrP did not affect shedding.
Andrew Thompson: Evaluating plasma tau and NfL as biomarkers for prion disease

Dr. Thompson opened by asking, after all the progress in prion disease biomarkers over the past several years, what do we still need? He sees three needs:
- Diagnostic markers for earlier diagnosis and for atypical subtypes.
- Quantitative markers of the disease process to be used as a clinical trial outcome measure.
- Marker of subclinical disease prior to symptom onset in people at risk.
His talk today will focus on 2 and 3. He selected total tau and NfL in plasma as good candidate biomakers for two reasons. First, because in many other neurological diseases, these exhibit promising properties, such as prodromal rise in Huntington’s disease [Byrne 2017] and response to treatment in multiple sclerosis [Novakova 2017]. Second, because these markers are already known to be elevated [Thompson 2018] and predictive [Staffaroni 2019] in symptomatic prion disease, as several groups have shown. (I’ve reviewed this literature in my fluid biomarkers post, and Thompson has reviewed it in [Thompson & Mead 2018].)
Dr. Thompson therefore looked at these markers in the UK National Prion Monitoring cohort, for all individuals with available plasma samples and appropriate consent. This included 231 sporadic CJD patients, 83 symptomatic genetic prion disease patients, and 23 pre-symptomatic genetic prion disease patients, of whom 9 converted to active disease. There were multiple samples over time available for many of the patients, so they had 709 plasma samples all told.
Among sporadic CJD patients, plasma NfL was categorically elevated above healthy controls, and correlated with score on the MRC Rating Scale [Thompson 2013]. In serial samples in symptomatic patients, plasma NfL showed a clear increase over time as the disease progressed. Plasma tau was also elevated and appeared to rise, but the trend was less clear.
Among genetic patients, they grouped samples by four categories: >2 years before onset, ≤2 years before onset, early symptomatic (no functional impairment, MRC score = 20) and established disease with functional impairment (MRC score ≤19). Both markers were elevated in symptomatic patients, and plasma NfL was elevated in individuals ≤2 years before onset compared to those >2 years before onset. They also looked at serial samples in the 9 individuals who converted to active disease. Plasma tau showed some whiff of rising over time prior to onset, but not many pre-symptomatic samples were above the normal range and there was a lot of within-individual variability. Plasma NfL showed the more clear trend, with some of the 9 individuals exhibiting plasma NfL levels clearly above the normal range at one or more timepoints in the few years before onset, and rising sharply as onset approached.
In the Q&A, Sonia Vallabh asked whether the 9 individuals who converted had rapidly or slowly progressive PRNP mutations. Thompson replied that several of them had more slowly progressive mutations.
Simon Mead: Genetic risk factors for sporadic CJD: replication, expression, function
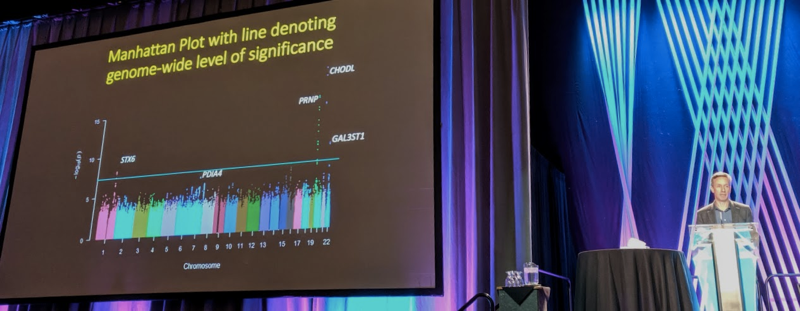
Dr. Mead began by motivating why to study genetic risk factors for prion disease. The hope is to use human genetics to dissect, of the many pathways that are reported to be involved in the initiation, spread, or replication of prions, which ones really matter. There is a reasonable prior for success because there has been success in finding genetic risk factors for other neurodegenerative diseases.
For 15 years now, Dr. Mead has been leading a consortium to aggregate DNA samples from sporadic CJD patients and perform genome-wide association studies (GWAS) to identify genetic risk factors. The first two installments of this analysis [Mead 2009, Mead 2012] did not identify any genome-wide signficant (P < 5 × 10-8) hits outside of PRNP, though they were suggestive that other risk factors might exist. In the new installment, there are genome-wide significant hits.
The discovery dataset consisted of N=4,110 CJD cases, most of them autopsy-confirmed, and >13,000 controls. All are of European ancestry. Principal components analysis suggested cases and controls were well-matched. In a Q-Q plot they had a genomic inflation factor (λ) of 1.04, which is considered acceptable (as a rule of thumb it should be <1.1).
As expected, PRNP was a strong hit. There were also a small number of SNPs with strong P values at CHODL, but that they had some QC issues and they now think this hit is not real. There are two other hits that are genome-wide significant (P < 5 × 10-8), and had real-looking linkage peaks. With the caveat that one cannot guarantee that the gene where lead SNPs are physically located is the causal gene, these SNPs were located at STX6 (odds ratio 1.19) and GAL3ST1 (odds ratio 1.17). There was also a suggestive hit at PDIA4 that did not quite cross the genome-wide significance threshold. In a replication dataset with another 962 cases and many more controls, they found strong independent replication for STX6, suggestive replication of GAL3ST1, and no particular supporting evidence for PDIA4 although the P value did get a little bit lower in the combined analysis, dropping into the 10-7 range.
Gene pathway analysis was not informative. None of the hits was correlated with PRNP codon 129 genotype (and by implication, PrPSc type 1 vs. 2, which is highly correlated with codon 129).
STX6 encodes a trans-Golgi protein that is a component of the SNARE complex involved in vesicle fusion. It is widely expressed throughout the body but especially high in cerebellum. The risk allele of the lead SNP is a GTEx cis-eQTL associated with higher expression of STX6 in cerebellum. In the UK National Prion Monitoring Cohort, STX6 genotype was not associated with survival time or rate of disease progression. The same risk haplotype has also been identified as a risk factor in GWAS for the tauopathy progressive supranuclear palsy (PSP) [Hoglinger 2011].
One of the lead SNPs for PDIA4 is also a cis-eQTL in brain. The lead SNPs for GAL3ST1 are not known to be associated with gene expression.
They also calculated a polygenic risk score (PRS) using genome-wide data. The PRS can identify 8% of the population with an odds ratio of 3 for elevated risk of CJD; this 8% of the population would include 21% of CJD cases. But for such a rare disease, this is not a strong enough enrichment to be clinically useful.
Dr. Mead is undertaking studies to determine if the genes they’ve identified are indeed the causal genes at these loci and, if so, to decipher the mechanism by which these genes control risk.
Andrew Reidenbach: NMR fragment screening to discover low molecular weight prion protein binders
Andrew works on our team at the Broad Institute and for the past 1.5 years has been principally responsible for our efforts to identify small molecules that bind PrP, while Sonia and I have been focused largely on ASOs (see above). He presented a poster on using NMR fragment screening to try to identify low molecular weight (<350 Da) binders of PrP. This is a sensitive approach capable of detecting even very weak binders, as starting points for structure-based drug design to identify more potent binders.
It turns out that finding PrP binders is really hard! Over the past few years and across several different fragment libraries, Andrew and I have screened a total of N=6,631 unique fragments, and only found one validated hit. Andrew has done a lot to try to invalidate it, but the hit appears to be real: it is dose-responsive, it exhibits a structure-activity relationship, and it does not seem to be an aggregator. But, it’s very weak (Kd > 1 mM), and the PrP residues that appear to be involved in the binding based on NMR shifts are not clustered tightly in one binding pocket, so we haven’t yet been able to fully characterize the exact binding site or mode. This hit could provide a starting point to develop a more potent binder, although the weak starting Kd is a big challenge. Andrew’s data also suggest that PrP is a difficult drug target for small molecules.
Chuck Hutti: Global analysis of protein degradation rates in prion infected cells by dynamic SILAC

Chuck Hutti is a PhD student in Sina Ghaemmaghami’s lab at University of Rochester. He presented a study of protein degradation rates in prion infected and non-infected cells using dynamic SILAC and mass spectrometry. The comparison was between RML prion-infected ScN2a cells, versus previously RML prion-infected ScN2a cells that had been cured of prion infection by treatment with quinacrine, an antiprion compound. Quinacrine itself is an inhibitor of lysosome function, and so would be expected to affect protein degradation, but Chuck clarified that the quinacrine treatment was transient and had washed out long before the cells were harvested for these experiments. They also replicated the results in N2a cells that had never been exposed to prions, providing further confidence that the quinacrine was not a confounder.
As you’d expect, they found that the degradation rate of PrP was slower in infected than non-infected cells, consistent with longstanding evidence that PrPSc has a longer half-life than PrPC [Safar 2005]. Interestingly, in order to make this comparison, they had to halt cell division with sodium butyrate. If you do the comparison in continuously dividing cells, you find only a smaller difference in PrP degradation rates, because dilution due to cell division is the predominant force that keeps PrPSc accumulation in check [Ghaemmaghami 2007]. When they looked, proteome-wide, at protein degradation rates they found that on average, proteins were degraded faster in infected cells. The increase in degradation rates was observed both for proteasome and lysosome substrates, and across many different categories of proteins, but not for components of the ribosome.
This result seems very surprising — wouldn’t you think that PrPSc accumulation in lysosomes might impair protein degradation? But on the other hand, it is known that both Shadoo and PrPC are post-translationally downregulated in prion-infected brains at end stage [Watts 2011, Mays 2014], and it has been hypothesized this reflects a cellular response to PrPSc of upregulating protein degradation — so you could argue the results here aren’t so strange after all. Still, I found it surprising and interesting.
These were a rushed few days — I missed a lot of talks and there were several poster presenters I wanted to chat with and didn’t get to. And we fly back to Boston tomorrow so will miss the whole last day. It’s been a wonderful few days of seeing so many colleagues and collaborators and I apologize to everyone I didn’t have more time to talk to! I also want to say thanks to everyone who allowed me to blog their presentations over these couple of days. It has been great seeing so many people in our research community this week, and we are grateful for all that you do.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.