A glimpse of non-PRNP genetic risk factors for prion disease
Last month, a vast international consortium led by Simon Mead pre-printed the first genome-wide association study (GWAS) to convincingly identify genetic risk factors for sporadic prion disease other than the prion protein gene itself [Jones 2020]. This study was long in the making — I first blogged about it six years ago in Trieste, Italy, when Simon was seeking out collaborators to contribute additional DNA samples. The reward for this incredible work of patience is a sample size that far outstrips that of previous GWAS in prion disease [Mead 2009, Mead 2012, Sanchez-Juan 2012, Sanchez-Juan 2015]:
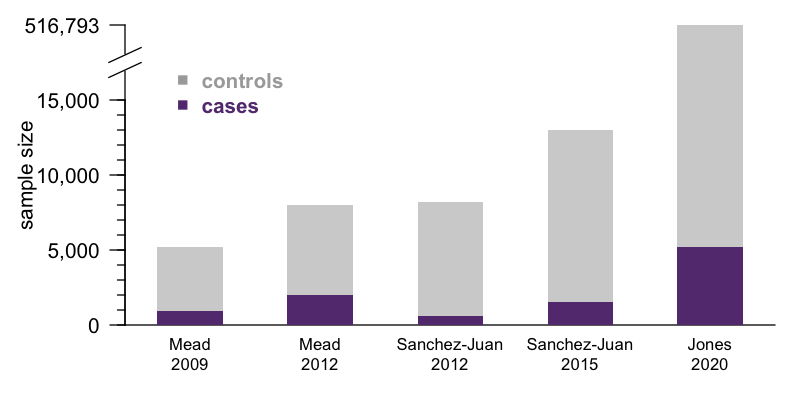
Indeed, this is likely to be the largest genetic association study we’ll see in prion disease for a long time. In more common diseases like heart disease or type 2 diabetes, the patients are out there, and GWAS sample size is limited only the ability to consent and enroll and genotype them and analyze the data. This ability is in turn a function of funding, consortium politics, personal motivation on the part of lead investigators. In prion disease, those are surely factors as well, but with 5,208 cases in this new study, we may be coming up against the limit of how many patients have ever been ascertained by the medical system. The number of confirmed cases is surely in the tens of thousands, but once you account for the criteria of needing research consents, definite or probable diagnosis, DNA availabilty, European ancestry, and so on, there may not be too many more eligible cases out there today. Getting a significantly larger sample size than this will require either waiting several years, or prion surveillance in China expanding enough to enable a comparable effort in the East Asian population.
It’s worth pointing out and emphasizing that, up to now, we have had no insight into any genetic risk factors other than the prion protein gene (PRNP) itself. There have been a handful of families worldwide with more than 1 case of prion disease but no PRNP mutation, but no more than would be expected by chance, and no other causal gene was ever mapped. The prior GWAS in this area [Mead 2009, Mead 2012, Sanchez-Juan 2012, Sanchez-Juan 2015] sometimes nominated suggestive associations, but none demonstrated statistically robust association to any other genes. Genetic studies in mice — QTL mapping and candidate gene knockout and transgenesis — never revealed anything robust and convincing either. Up to now, there has simply been nothing except PRNP.
The new study finds two hit loci, at 1q25.3 and 22q12.2. In terms of statistics and quality control, both hits look real. They exceed the genome-wide significance threshold (lead SNPs P = 9.7e-9 and 8.6e-10 respectively). They have sensible-looking linkage disequilibrium peaks, arguing against a technical artifact. And the genomic inflation factor is just 1.026, meaning the cases and controls were well-matched, so a population stratification confounder is unlikely.
In general, mapping GWAS loci to causal genes is often really hard. In a different version of history, we could have gotten two clear hit loci for prion disease risk and still been years — someone’s whole postdoc — away from knowing which genes actually control risk. In this case, it looks like we got lucky: the two loci each seem to map fairly unambiguously onto one obvious gene. Statistical genetics skeptics might say that’s a strong claim, so I’ll take a brief detour here to unpack the evidence for causal gene assignment.
The lead SNP for 1q25.3 is an eQTL (a genetic variant that controls gene expression) for the gene STX6, while the lead SNP for 22q12.2 is a missense variant in GAL3ST1. I pulled the variant-to-gene (v2g) mapping metrics from Open Targets Genetics for both leads and had a look. According to OTG’s scoring algorithm, these two are indeed the top-ranked genes for their respective lead SNPs, though neither is quite an open-and-shut case. The 1q25.3 SNP is an eQTL for STX6 but also for two genes located slightly further away, IER5 and MR1. STX6 is ranked 1st and commands a 20% share of all the overall v2g evidence assigned across genes. The 22q12.2 SNP is a missense variant for GAL3ST1 but also an eQTL for TCN2, DUSP18, and SEC14L6. GAL3ST1 is ranked 1st but still commands just 9% of the overall v2g share. Here are plots of v2g scoring at each locus:
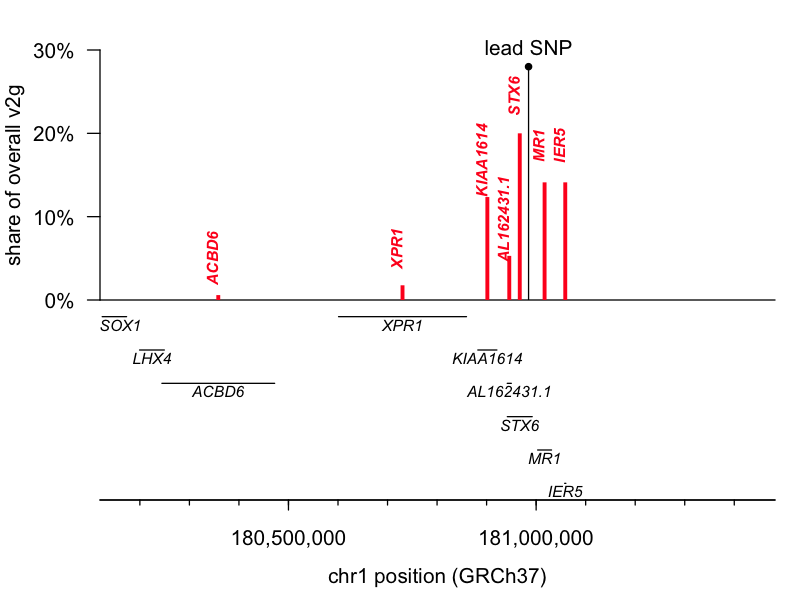
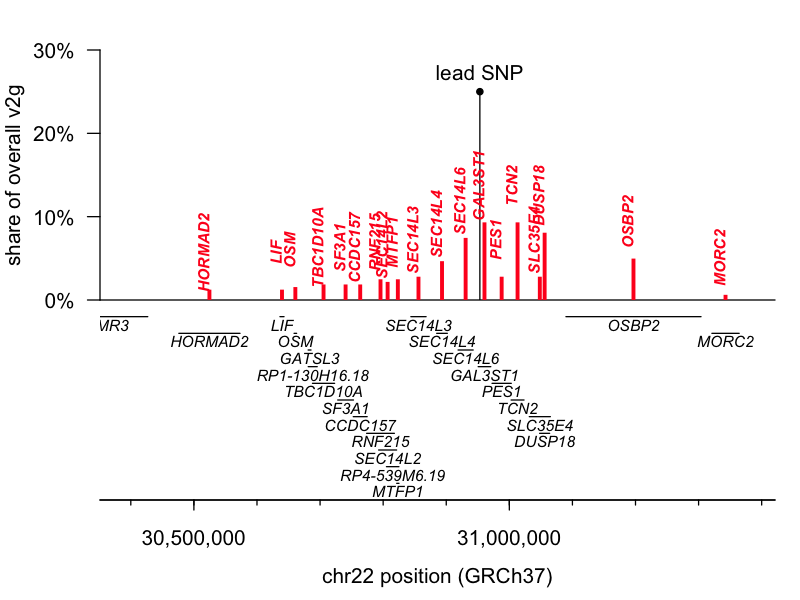
But, those data are based just on the known properties of the single lead SNP and do not take into account the full shape of the linkage disequilibrium peak at each locus. The authors further report performing colocalization analysis [Giambartolomei 2014], which revealed a tidy overlap between the peak for prion disease risk and the peak for STX6 expression, further supporting this causal gene. Moreover, while we must always be open to novel biology — say, involvement of a tissue we didn’t think was important in disease — biological plausibility also has some role in setting our priors. According to the GTEx portal, the 1q lead SNP is an eQTL for STX6 expression in the brain, where we expect it could reasonably matter, while it affects MR1 expression only in skeletal muscle, where a connection to prion disease seems far less likely. Similarly, the GTEx portal shows the 22q SNP affecting TCN2 and SEC14L6 expression in tissues like skin, muscle, and esophagus.
Overall, then, my take is that the authors are correct — we can be fairly confident that STX6 and GAL3ST1 are the causal genes at these two hit loci.
Why do we care about identifying new genetic risk factors in prion disease? A few potential reasons. Sometimes GWAS can shed light on fundamental biology, for example, in schizophrenia, where until GWAS we really had little insight into what the nature of the disease even was [Sekar 2016]. Sometimes GWAS can predict who is at risk, for example in heart disease, where polygenic risk score can identify people at just as high of risk as that conferred by monogenic mutations [Khera 2018]. Sometimes GWAS can point to new drug targets, which will have an outsize chance of clinical success [Nelson 2015, King 2019]. Let’s visit each of these topics in turn.
In terms of fundamental biology, I’d say we haven’t learned much yet, but there exists potential. STX6 encodes syntaxin-6, a component of the t-SNARE complex involved in endosomal transport. There was already some chemical biology evidence for the importance of endosomal recycling in prion disease, and this hit could turn out to provide human genetic validation to back it up. But beyond that, it’s early days. We don’t know in much more granular detail exactly what syntaxin-6 does or why it might matter in prion disease. Hopefully identification of this causal gene opens the door to functional studies that will reveal more, but extracting real insights will take years. GAL3ST1 encodes galactose-3-o-sulfotransferase 1, an enzyme involved in producing a specific type of lipid found in myelin. PrP’s native function, at least in peripheral nerves, has something to do with myelin maintenance, so one can imagine all sorts of biological connections, but again, to say in any more granular detail why this enzyme matters will take years.
As an aside, does the identification of new risk genes mean that prion disease is now a polygenic disease? We could always choose to get lost in semantics here, but I’m going to argue no, it doesn’t. By definition, if someone doesn’t have misfolded PrP in their brain, it’s not prion disease. The single gene PRNP still encodes the sole causal protein. Based on everything we know so far about prion disease biology, we expect that other genes that affect risk could only do so through PrP — though proving this and figuring out how, exactly, will be a long road. For now, my take-home is that prion disease is still monogenic in the sense that a single protein causes all cases of the disease.
In terms of risk prediction, I’d say there’s not much value here, and there never will be, because prion disease is simply too rare (and I believe the authors share this view). Let’s dive into the numbers for a moment. Prion disease kills about 1 in every 6,000 people [Maddox 2020], a baseline risk of about 0.017%. Both new associations are — as one expects for GWAS signals — modest, with odds ratios of 1.14 and 1.11 in an allelic model. That means that if you were homozygous for the risk alleles for both genes, your lifetime risk would rise to about 0.027%. That’s not exactly actionable information. Now, polygenic risk scoring allows us to use genome-wide information to predict a person’s risk, without being limited to genome-wide significant loci. But this study found the heritability of prion disease risk to be only about 25-26%. That’s a lot less than, say, height, which is ~80% heritable [Wainschtein 2019]. For a binary trait like prion disease (yes you have it or no you don’t), it’s hard to put into words what exactly heritability means. Perhaps the best way to explain the limitation here is the calculation that Dr. Mead shared at Prion2019. He said that a genome-wide polygenic risk score based on his GWAS could identify 8% of the population that is at 3x higher risk of prion disease than everyone else; these 8% of people would consist of 21% of cases. This, as he pointed out, is not clinically useful. Of course, for a common condition like heart attack, such enrichment can be quite clinically useful [Khera 2018], but for a disease that will only ever affect 0.02% of us, even one day when we have a preventive drug in hand, the cost, invasiveness, and inconvenience of screening 8% of the population would appear untenable.
If these specific hits, or even polygenic risk scores, appear not to be useful for predicting sporadic prion disease risk, one might still ask if they could be useful in genetic prion disease. Among people with high-penetrance variants, at ≥90% risk of developing prion disease, could a risk score help to predict age of onset? There could be some predictive value there, but I would be surprised if it rises to the level of being clinically meaningful. My skepticism stems from the fact that even the common M129V polymorphism in PRNP, which powerfully controls so many aspects of prion disease risk and clinical presentation, appears to have almost no predictive value for age onset in genetic prion disease [Minikel 2019]. Indeed, even which mutation you have in PRNP barely has predictive value: the mean ages of onset are significantly different, but the ranges are wide and overlapping, so that mutation explains only about 15% of variance [Minikel 2019]. If even PRNP can’t predict age of onset well, it seems somewhat unlikely that a polygenic risk score could get us to a point where it would improve genetic counseling or allow us to design better clinical trials.
In terms of identifying potential drug targets, I view this work as an important step forward, but for now it’s not going to change my day-to-day priorities in the lab. Based on the data presented here, I would now rank STX6 and GAL3ST1 as the distant second and third best drug targets in prion disease, after PRNP. Let me unpack that claim in two more paragaphs.
On one hand, I rank these targets so high because I believe human genetics has excellent model validity. It’s the species we care about, it’s in vivo, whole-organism data, and the statistical methods employed in GWAS are rigorous and well-controlled, severely limiting false positives. In this case, the variant-to-gene mapping also appears strong. Empirically, drug discovery programs with targets backed by human genetic support are more likely to succeed [Nelson 2015, King 2019]. In addition, there are other things potentially appealing about each of these targets. GAL3ST1 encodes an enzyme, and is thus a canonically “druggable” type of target. STX6 is also a GWAS hit for tauopathy risk [Hoglinger 2011, Ferrari 2014], and with the same direction of effect — the allele that increases STX6 expression also increases risk for both diseases — so this might provide a rare opportunity for a therapeutic hypothesis relevant across more than one form of dementia. For all these reasons, someone out there may want to launch a drug discovery campaign against these targets, and if they do, I’d be thrilled.
At the same time, I say that I rank these targets rather distantly after PRNP because they are surely less central to the disease process, and there is so much we still don’t know about their role. A pivotal question is, do these genes affect the rate at which prions replicate, the rate at which they cause neurotoxicity — or only the probability of that first prion forming in the brain to begin with? The paper provides a bit of evidence for the latter. Genotypes at neither gene were associated with rate of disease progression, survival, or any other clinical phenotypes in the subset of individuals with these data points available. (As a positive control, PRNP genotype was correlated with many of these outcomes). Knockdown of syntaxin-6 also did not affect prion accumulation in cell culture. If these genes only affect initiation, and not progression, of prion disease, then they might not be plausible targets in the symptomatic phase of disease. They might be highly effective targets to prevent disease at a presymptomatic stage, but, unlike PRNP, they might not lend themselves to any obvious biomarker readout to confirm that the drug is doing anything, and their presumably less central role would make them less strong candidates for a surrogate endpoint strategy. In short, we don’t yet know at what disease stage these targets are relevant, or how one could design a clinical drug development program around them. And these are just a couple of key items in a whole checklist of other things one wants in hand when designing a drug discovery program: pharmacodynamic biomarker, dose-response relationship, established relevance of target in animal models and/or existence of humanized animal models, and so on. These are all boxes we can now check for PRNP, and not a moment too soon, 35 years after this molecular target first came into view. STX6 and GAL3ST1 have only now just come into the picture, and I believe this is an important development, but for the foreseeable future, my focus will remain on the target we know: PRNP.
All that being said, there is no doubt this study is an important step forward, a true labor of love years in the making, and I extend kudos to the investigators who led it, all the consortium members who contributed, and above all, the patients who donated their data to research.

About Eric Vallabh Minikel
Eric Vallabh Minikel is on a lifelong quest to prevent prion disease. He is a scientist based at the Broad Institute of MIT and Harvard.